 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeConformal Prediction Sets with Limited False Positives
Feb 15, 2022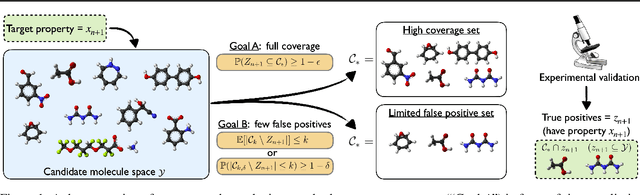
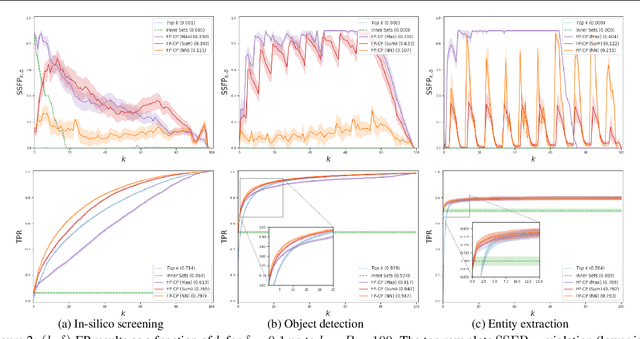
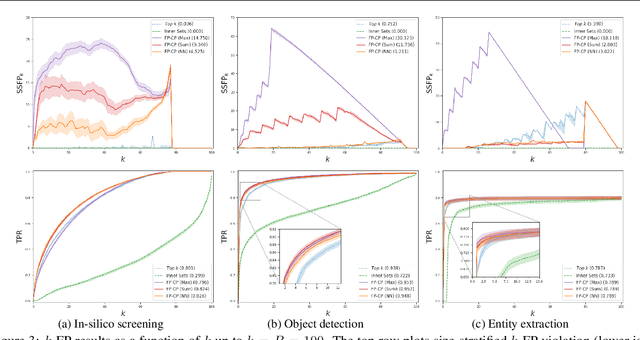
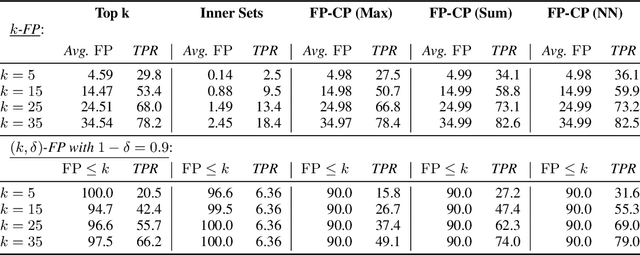
We develop a new approach to multi-label conformal prediction in which we aim to output a precise set of promising prediction candidates with a bounded number of incorrect answers. Standard conformal prediction provides the ability to adapt to model uncertainty by constructing a calibrated candidate set in place of a single prediction, with guarantees that the set contains the correct answer with high probability. In order to obey this coverage property, however, conformal sets can become inundated with noisy candidates -- which can render them unhelpful in practice. This is particularly relevant to practical applications where there is a limited budget, and the cost (monetary or otherwise) associated with false positives is non-negligible. We propose to trade coverage for a notion of precision by enforcing that the presence of incorrect candidates in the predicted conformal sets (i.e., the total number of false positives) is bounded according to a user-specified tolerance. Subject to this constraint, our algorithm then optimizes for a generalized notion of set coverage (i.e., the true positive rate) that allows for any number of true answers for a given query (including zero). We demonstrate the effectiveness of this approach across a number of classification tasks in natural language processing, computer vision, and computational chemistry.
EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction
Feb 07, 2022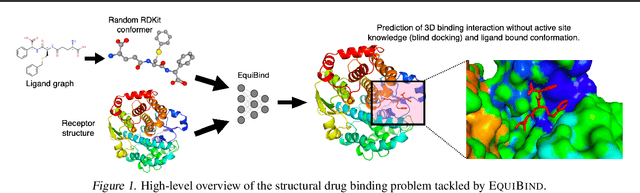
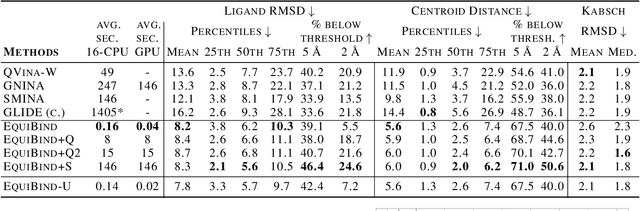
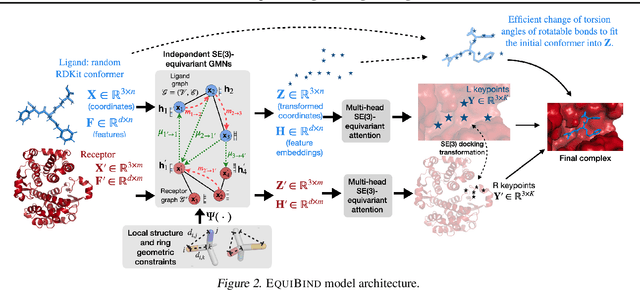
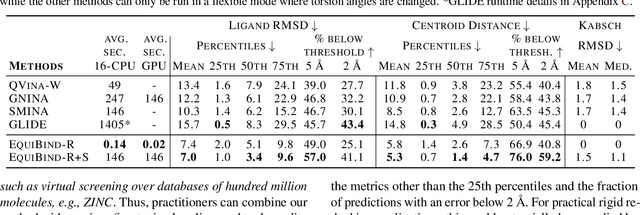
Predicting how a drug-like molecule binds to a specific protein target is a core problem in drug discovery. An extremely fast computational binding method would enable key applications such as fast virtual screening or drug engineering. Existing methods are computationally expensive as they rely on heavy candidate sampling coupled with scoring, ranking, and fine-tuning steps. We challenge this paradigm with EquiBind, an SE(3)-equivariant geometric deep learning model performing direct-shot prediction of both i) the receptor binding location (blind docking) and ii) the ligand's bound pose and orientation. EquiBind achieves significant speed-ups and better quality compared to traditional and recent baselines. Further, we show extra improvements when coupling it with existing fine-tuning techniques at the cost of increased running time. Finally, we propose a novel and fast fine-tuning model that adjusts torsion angles of a ligand's rotatable bonds based on closed-form global minima of the von Mises angular distance to a given input atomic point cloud, avoiding previous expensive differential evolution strategies for energy minimization.
Controlling Directions Orthogonal to a Classifier
Jan 27, 2022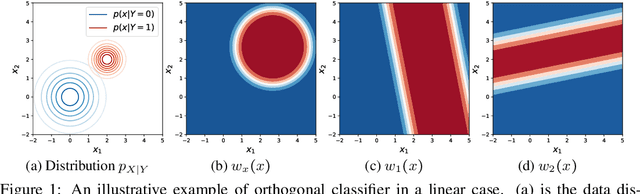
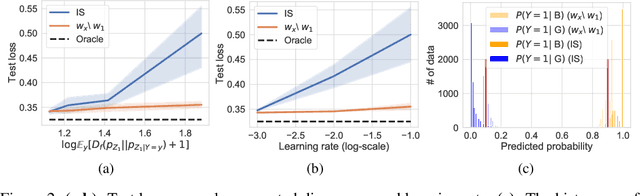


We propose to identify directions invariant to a given classifier so that these directions can be controlled in tasks such as style transfer. While orthogonal decomposition is directly identifiable when the given classifier is linear, we formally define a notion of orthogonality in the non-linear case. We also provide a surprisingly simple method for constructing the orthogonal classifier (a classifier utilizing directions other than those of the given classifier). Empirically, we present three use cases where controlling orthogonal variation is important: style transfer, domain adaptation, and fairness. The orthogonal classifier enables desired style transfer when domains vary in multiple aspects, improves domain adaptation with label shifts and mitigates the unfairness as a predictor. The code is available at http://github.com/Newbeeer/orthogonal_classifier
Independent SE-Equivariant Models for End-to-End Rigid Protein Docking
Nov 15, 2021



Protein complex formation is a central problem in biology, being involved in most of the cell's processes, and essential for applications, e.g. drug design or protein engineering. We tackle rigid body protein-protein docking, i.e., computationally predicting the 3D structure of a protein-protein complex from the individual unbound structures, assuming no conformational change within the proteins happens during binding. We design a novel pairwise-independent SE(3)-equivariant graph matching network to predict the rotation and translation to place one of the proteins at the right docked position relative to the second protein. We mathematically guarantee a basic principle: the predicted complex is always identical regardless of the initial locations and orientations of the two structures. Our model, named EquiDock, approximates the binding pockets and predicts the docking poses using keypoint matching and alignment, achieved through optimal transport and a differentiable Kabsch algorithm. Empirically, we achieve significant running time improvements and often outperform existing docking software despite not relying on heavy candidate sampling, structure refinement, or templates.
Fragment-based Sequential Translation for Molecular Optimization
Oct 26, 2021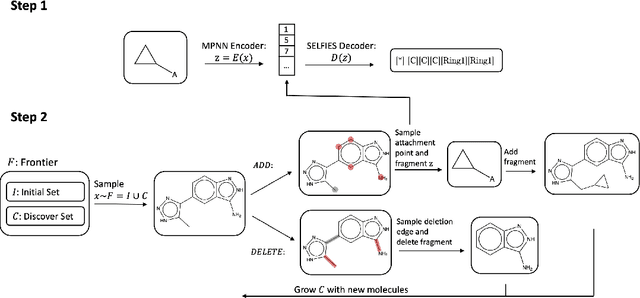
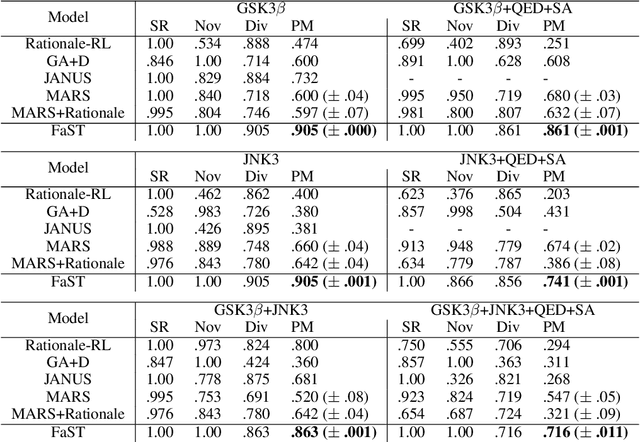
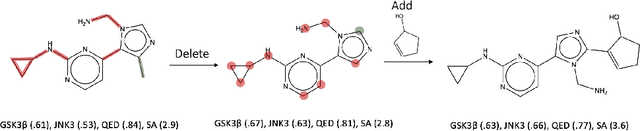
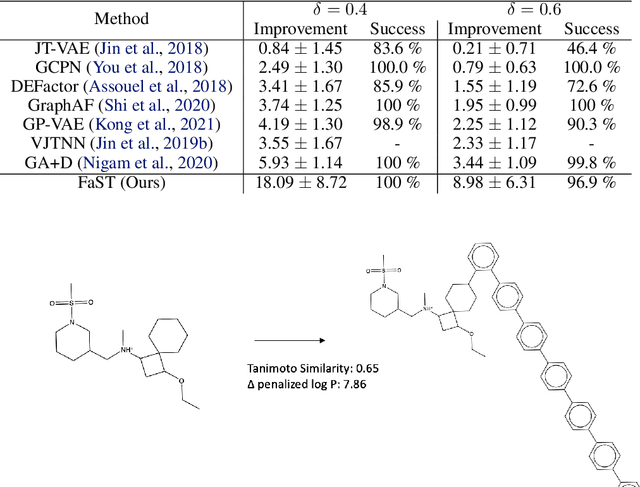
Searching for novel molecular compounds with desired properties is an important problem in drug discovery. Many existing frameworks generate molecules one atom at a time. We instead propose a flexible editing paradigm that generates molecules using learned molecular fragments--meaningful substructures of molecules. To do so, we train a variational autoencoder (VAE) to encode molecular fragments in a coherent latent space, which we then utilize as a vocabulary for editing molecules to explore the complex chemical property space. Equipped with the learned fragment vocabulary, we propose Fragment-based Sequential Translation (FaST), which learns a reinforcement learning (RL) policy to iteratively translate model-discovered molecules into increasingly novel molecules while satisfying desired properties. Empirical evaluation shows that FaST significantly improves over state-of-the-art methods on benchmark single/multi-objective molecular optimization tasks.
Learning Representations that Support Robust Transfer of Predictors
Oct 19, 2021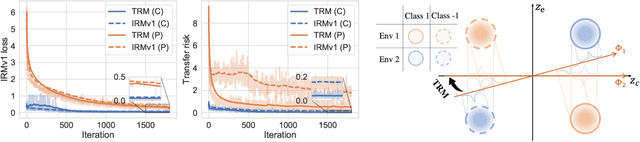
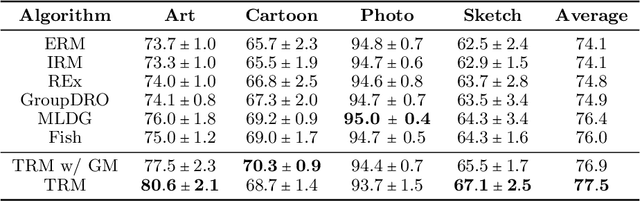
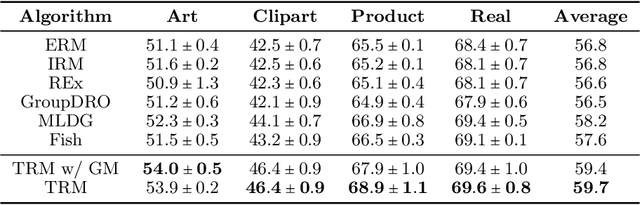

Ensuring generalization to unseen environments remains a challenge. Domain shift can lead to substantially degraded performance unless shifts are well-exercised within the available training environments. We introduce a simple robust estimation criterion -- transfer risk -- that is specifically geared towards optimizing transfer to new environments. Effectively, the criterion amounts to finding a representation that minimizes the risk of applying any optimal predictor trained on one environment to another. The transfer risk essentially decomposes into two terms, a direct transfer term and a weighted gradient-matching term arising from the optimality of per-environment predictors. Although inspired by IRM, we show that transfer risk serves as a better out-of-distribution generalization criterion, both theoretically and empirically. We further demonstrate the impact of optimizing such transfer risk on two controlled settings, each representing a different pattern of environment shift, as well as on two real-world datasets. Experimentally, the approach outperforms baselines across various out-of-distribution generalization tasks. Code is available at \url{https://github.com/Newbeeer/TRM}.
Iterative Refinement Graph Neural Network for Antibody Sequence-Structure Co-design
Oct 15, 2021
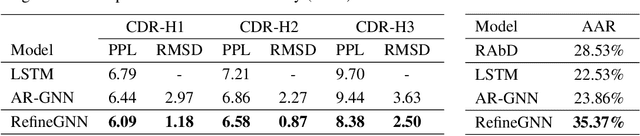
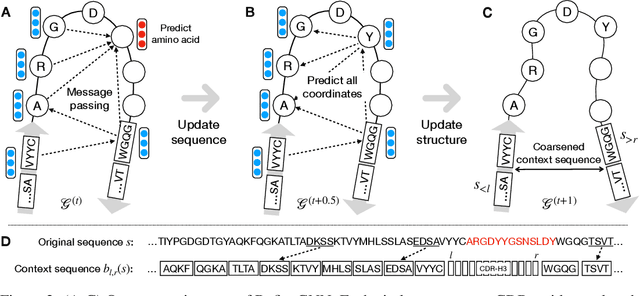
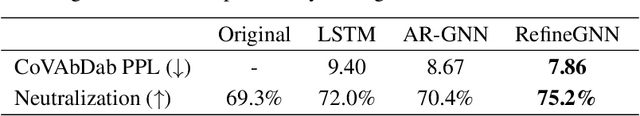
Antibodies are versatile proteins that bind to pathogens like viruses and stimulate the adaptive immune system. The specificity of antibody binding is determined by complementarity-determining regions (CDRs) at the tips of these Y-shaped proteins. In this paper, we propose a generative model to automatically design the CDRs of antibodies with enhanced binding specificity or neutralization capabilities. Previous generative approaches formulate protein design as a structure-conditioned sequence generation task, assuming the desired 3D structure is given a priori. In contrast, we propose to co-design the sequence and 3D structure of CDRs as graphs. Our model unravels a sequence autoregressively while iteratively refining its predicted global structure. The inferred structure in turn guides subsequent residue choices. For efficiency, we model the conditional dependence between residues inside and outside of a CDR in a coarse-grained manner. Our method achieves superior log-likelihood on the test set and outperforms previous baselines in designing antibodies capable of neutralizing the SARS-CoV-2 virus.
Crystal Diffusion Variational Autoencoder for Periodic Material Generation
Oct 12, 2021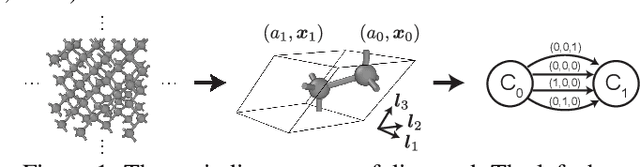

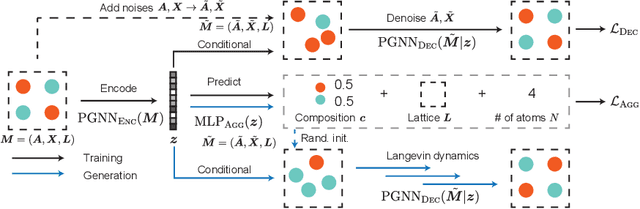
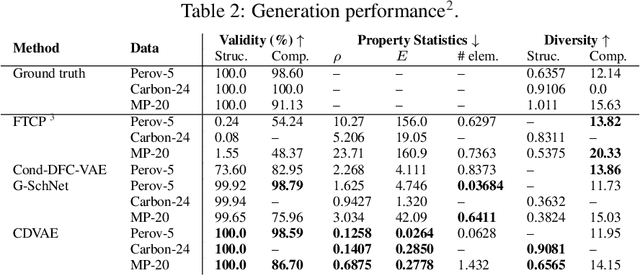
Generating the periodic structure of stable materials is a long-standing challenge for the material design community. This task is difficult because stable materials only exist in a low-dimensional subspace of all possible periodic arrangements of atoms: 1) the coordinates must lie in the local energy minimum defined by quantum mechanics, and 2) global stability also requires the structure to follow the complex, yet specific bonding preferences between different atom types. Existing methods fail to incorporate these factors and often lack proper invariances. We propose a Crystal Diffusion Variational Autoencoder (CDVAE) that captures the physical inductive bias of material stability. By learning from the data distribution of stable materials, the decoder generates materials in a diffusion process that moves atomic coordinates towards a lower energy state and updates atom types to satisfy bonding preferences between neighbors. Our model also explicitly encodes interactions across periodic boundaries and respects permutation, translation, rotation, and periodic invariances. We significantly outperform past methods in three tasks: 1) reconstructing the input structure, 2) generating valid, diverse, and realistic materials, and 3) generating materials that optimize a specific property. We also provide several standard datasets and evaluation metrics for the broader machine learning community.
Learning Task Informed Abstractions
Jun 30, 2021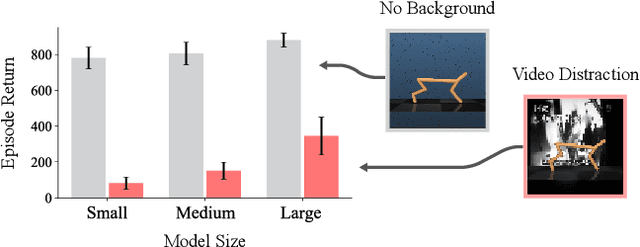

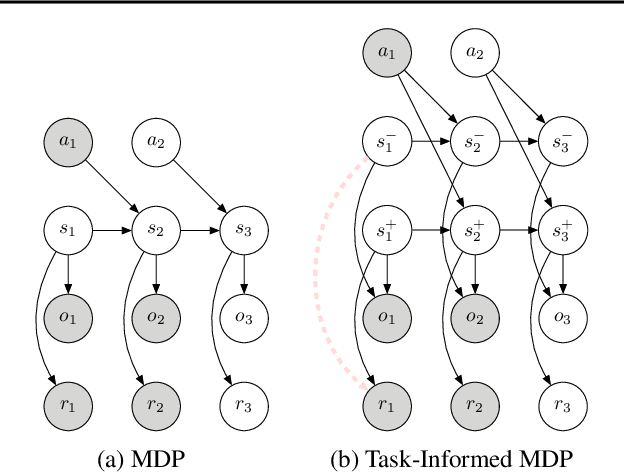
Current model-based reinforcement learning methods struggle when operating from complex visual scenes due to their inability to prioritize task-relevant features. To mitigate this problem, we propose learning Task Informed Abstractions (TIA) that explicitly separates reward-correlated visual features from distractors. For learning TIA, we introduce the formalism of Task Informed MDP (TiMDP) that is realized by training two models that learn visual features via cooperative reconstruction, but one model is adversarially dissociated from the reward signal. Empirical evaluation shows that TIA leads to significant performance gains over state-of-the-art methods on many visual control tasks where natural and unconstrained visual distractions pose a formidable challenge.
Consistent Accelerated Inference via Confident Adaptive Transformers
Apr 18, 2021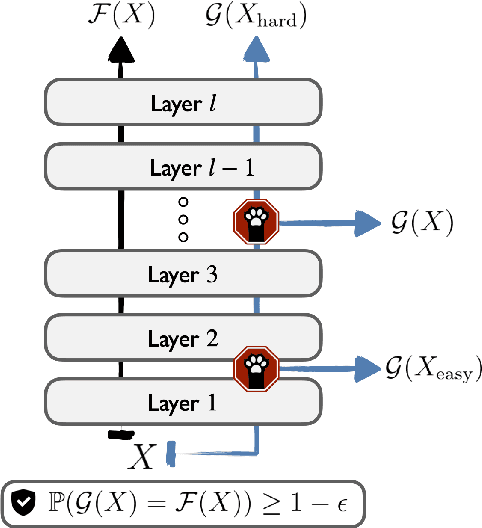

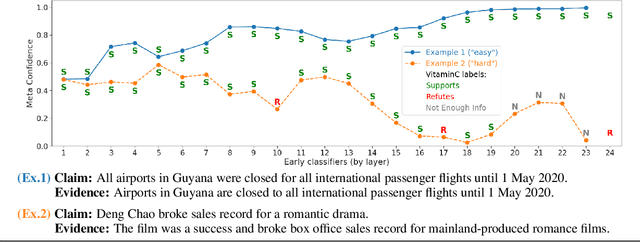
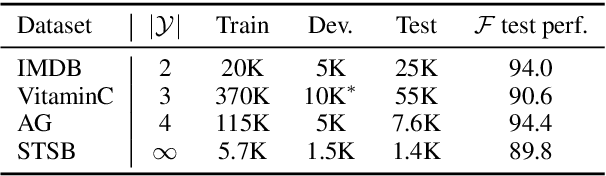
We develop a novel approach for confidently accelerating inference in the large and expensive multilayer Transformers that are now ubiquitous in natural language processing (NLP). Amortized or approximate computational methods increase efficiency, but can come with unpredictable performance costs. In this work, we present CATs -- Confident Adaptive Transformers -- in which we simultaneously increase computational efficiency, while guaranteeing a specifiable degree of consistency with the original model with high confidence. Our method trains additional prediction heads on top of intermediate layers, and dynamically decides when to stop allocating computational effort to each input using a meta consistency classifier. To calibrate our early prediction stopping rule, we formulate a unique extension of conformal prediction. We demonstrate the effectiveness of this approach on four classification and regression tasks.