 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccurate Machine Learned Quantum-Mechanical Force Fields for Biomolecular Simulations
May 17, 2022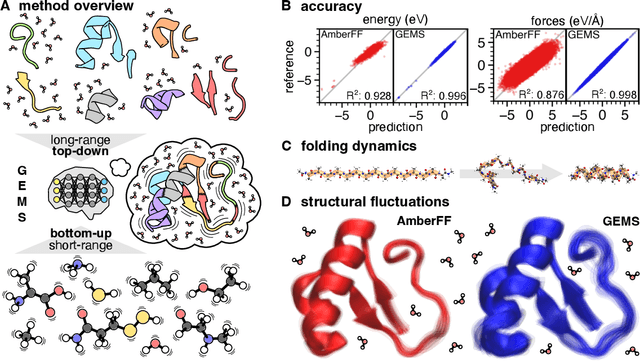
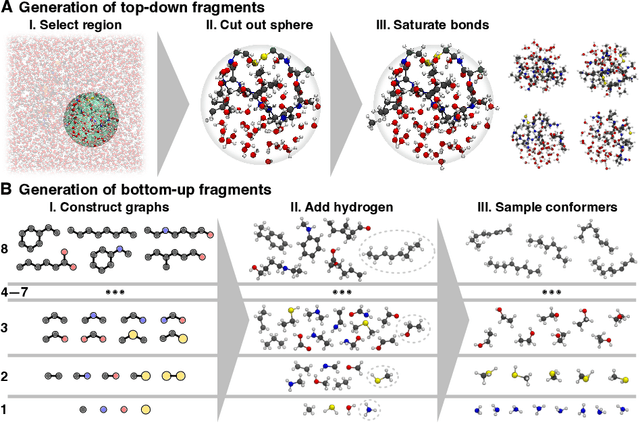
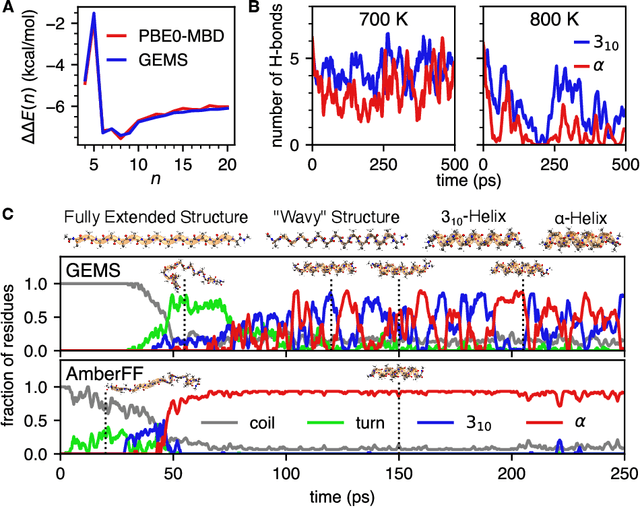
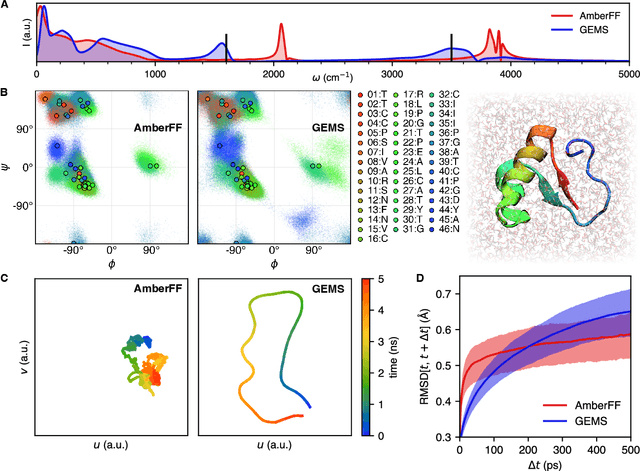
Molecular dynamics (MD) simulations allow atomistic insights into chemical and biological processes. Accurate MD simulations require computationally demanding quantum-mechanical calculations, being practically limited to short timescales and few atoms. For larger systems, efficient, but much less reliable empirical force fields are used. Recently, machine learned force fields (MLFFs) emerged as an alternative means to execute MD simulations, offering similar accuracy as ab initio methods at orders-of-magnitude speedup. Until now, MLFFs mainly capture short-range interactions in small molecules or periodic materials, due to the increased complexity of constructing models and obtaining reliable reference data for large molecules, where long-ranged many-body effects become important. This work proposes a general approach to constructing accurate MLFFs for large-scale molecular simulations (GEMS) by training on "bottom-up" and "top-down" molecular fragments of varying size, from which the relevant physicochemical interactions can be learned. GEMS is applied to study the dynamics of alanine-based peptides and the 46-residue protein crambin in aqueous solution, allowing nanosecond-scale MD simulations of >25k atoms at essentially ab initio quality. Our findings suggest that structural motifs in peptides and proteins are more flexible than previously thought, indicating that simulations at ab initio accuracy might be necessary to understand dynamic biomolecular processes such as protein (mis)folding, drug-protein binding, or allosteric regulation.
Variance Reduction in Deep Learning: More Momentum is All You Need
Nov 23, 2021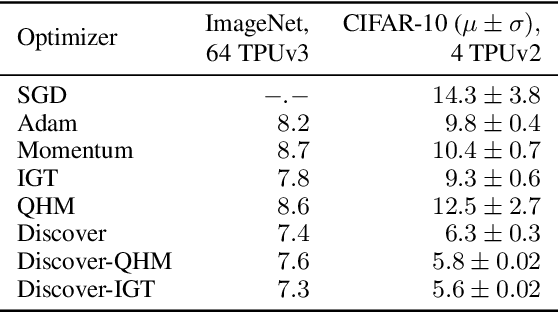
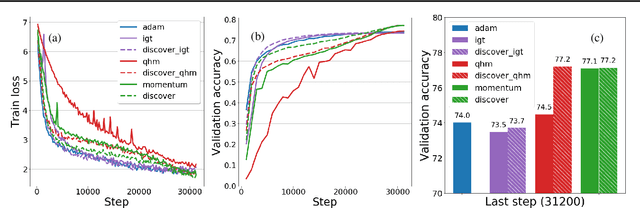
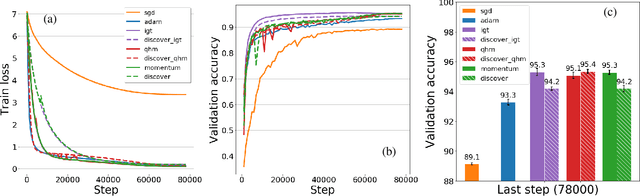
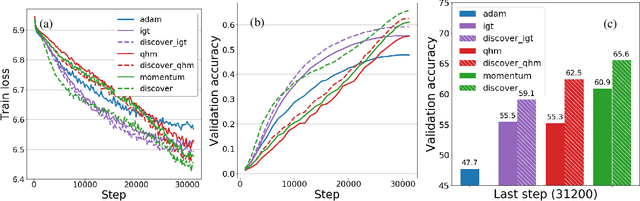
Variance reduction (VR) techniques have contributed significantly to accelerating learning with massive datasets in the smooth and strongly convex setting (Schmidt et al., 2017; Johnson & Zhang, 2013; Roux et al., 2012). However, such techniques have not yet met the same success in the realm of large-scale deep learning due to various factors such as the use of data augmentation or regularization methods like dropout (Defazio & Bottou, 2019). This challenge has recently motivated the design of novel variance reduction techniques tailored explicitly for deep learning (Arnold et al., 2019; Ma & Yarats, 2018). This work is an additional step in this direction. In particular, we exploit the ubiquitous clustering structure of rich datasets used in deep learning to design a family of scalable variance reduced optimization procedures by combining existing optimizers (e.g., SGD+Momentum, Quasi Hyperbolic Momentum, Implicit Gradient Transport) with a multi-momentum strategy (Yuan et al., 2019). Our proposal leads to faster convergence than vanilla methods on standard benchmark datasets (e.g., CIFAR and ImageNet). It is robust to label noise and amenable to distributed optimization. We provide a parallel implementation in JAX.
Continental-Scale Building Detection from High Resolution Satellite Imagery
Jul 29, 2021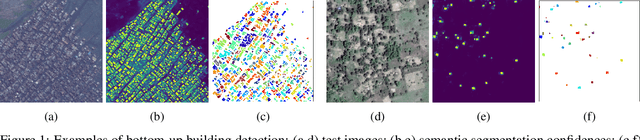
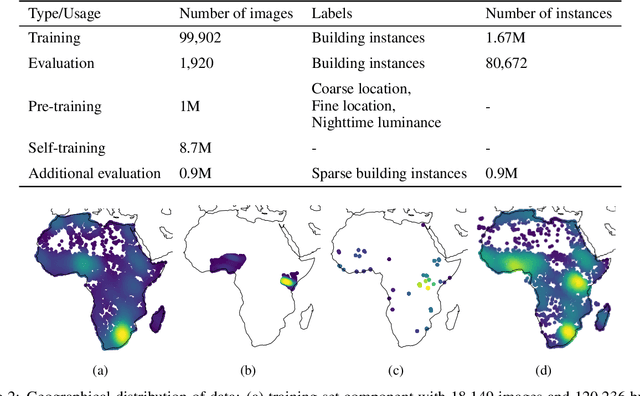

Identifying the locations and footprints of buildings is vital for many practical and scientific purposes. Such information can be particularly useful in developing regions where alternative data sources may be scarce. In this work, we describe a model training pipeline for detecting buildings across the entire continent of Africa, using 50 cm satellite imagery. Starting with the U-Net model, widely used in satellite image analysis, we study variations in architecture, loss functions, regularization, pre-training, self-training and post-processing that increase instance segmentation performance. Experiments were carried out using a dataset of 100k satellite images across Africa containing 1.75M manually labelled building instances, and further datasets for pre-training and self-training. We report novel methods for improving performance of building detection with this type of model, including the use of mixup (mAP +0.12) and self-training with soft KL loss (mAP +0.06). The resulting pipeline obtains good results even on a wide variety of challenging rural and urban contexts, and was used to create the Open Buildings dataset of 516M Africa-wide detected footprints.
Measuring Compositional Generalization: A Comprehensive Method on Realistic Data
Dec 20, 2019

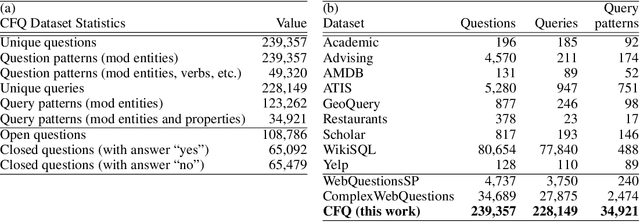
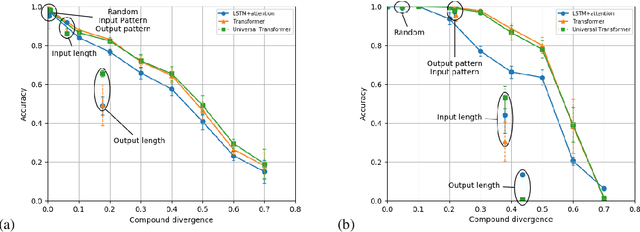
State-of-the-art machine learning methods exhibit limited compositional generalization. At the same time, there is a lack of realistic benchmarks that comprehensively measure this ability, which makes it challenging to find and evaluate improvements. We introduce a novel method to systematically construct such benchmarks by maximizing compound divergence while guaranteeing a small atom divergence between train and test sets, and we quantitatively compare this method to other approaches for creating compositional generalization benchmarks. We present a large and realistic natural language question answering dataset that is constructed according to this method, and we use it to analyze the compositional generalization ability of three machine learning architectures. We find that they fail to generalize compositionally and that there is a surprisingly strong negative correlation between compound divergence and accuracy. We also demonstrate how our method can be used to create new compositionality benchmarks on top of the existing SCAN dataset, which confirms these findings.