 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHarmonizable mixture kernels with variational Fourier features
Oct 11, 2018
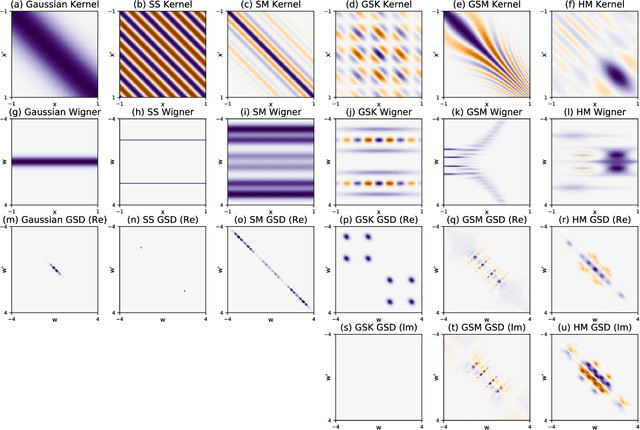
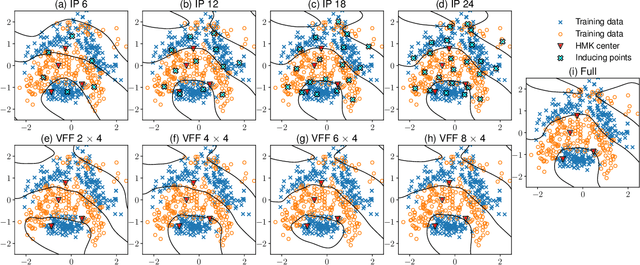
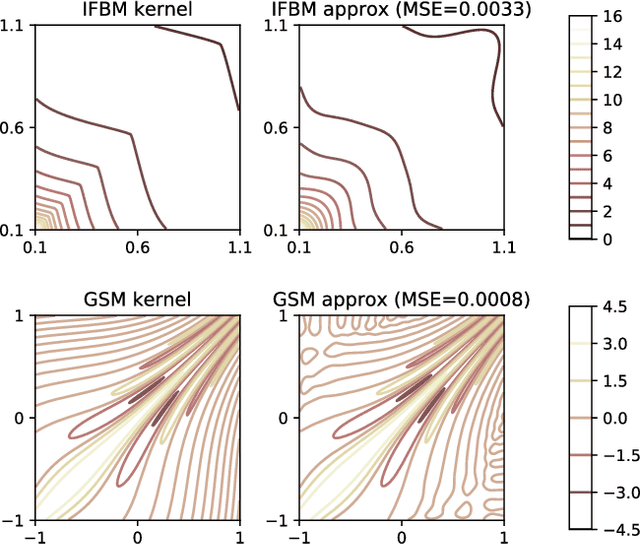
The expressive power of Gaussian processes depends heavily on the choice of kernel. In this work we propose the novel harmonizable mixture kernel (HMK), a family of expressive, interpretable, non-stationary kernels derived from mixture models on the generalized spectral representation. As a theoretically sound treatment of non-stationary kernels, HMK supports harmonizable covariances, a wide subset of kernels including all stationary and many non-stationary covariances. We also propose variational Fourier features, an inter-domain sparse GP inference framework that offers a representative set of 'inducing frequencies'. We show that harmonizable mixture kernels interpolate between local patterns, and that variational Fourier features offers a robust kernel learning framework for the new kernel family.
Deep convolutional Gaussian processes
Oct 06, 2018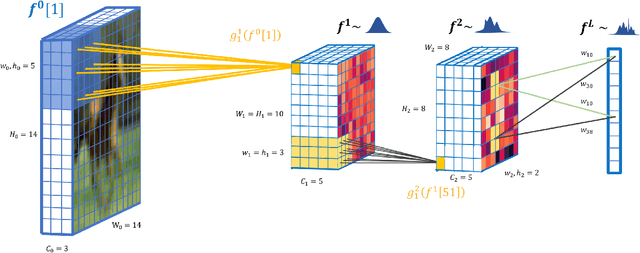
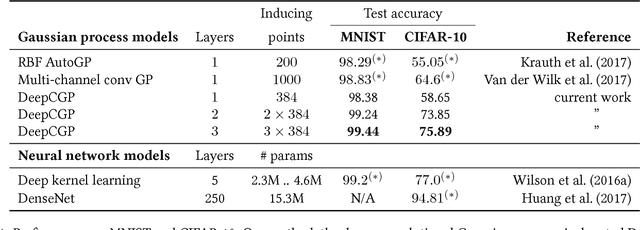
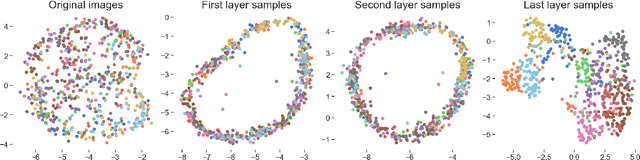
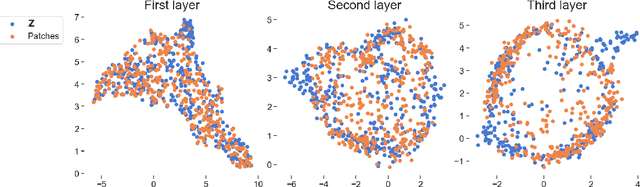
We propose deep convolutional Gaussian processes, a deep Gaussian process architecture with convolutional structure. The model is a principled Bayesian framework for detecting hierarchical combinations of local features for image classification. We demonstrate greatly improved image classification performance compared to current Gaussian process approaches on the MNIST and CIFAR-10 datasets. In particular, we improve CIFAR-10 accuracy by over 10 percentage points.
Modelling User's Theory of AI's Mind in Interactive Intelligent Systems
Sep 08, 2018
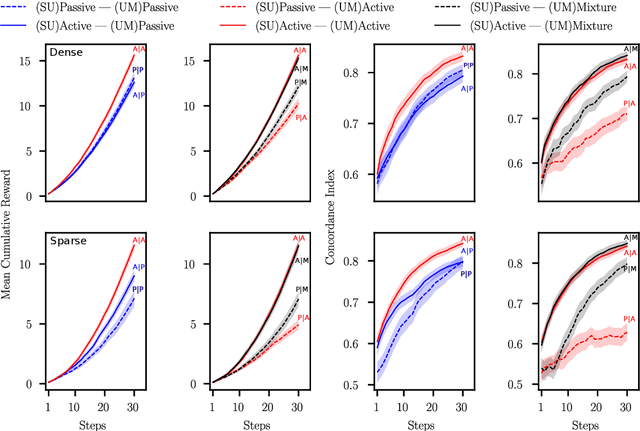

Many interactive intelligent systems, such as recommendation and information retrieval systems, treat users as a passive data source. Yet, users form mental models of systems and instead of passively providing feedback to the queries of the system, they will strategically plan their actions within the constraints of the mental model to steer the system and achieve their goals faster. We propose to explicitly account for the user's theory of the AI's mind in the user model: the intelligent system has a model of the user having a model of the intelligent system. We study a case where the system is a contextual bandit and the user model is a Markov decision process that plans based on a simpler model of the bandit. Inference in the model can be reduced to probabilistic inverse reinforcement learning, with the nested bandit model defining the transition dynamics, and is implemented using probabilistic programming. Our results show that improved performance is achieved if users can form accurate mental models that the system can capture, implying predictability of the interactive intelligent system is important not only for the user experience but also for the design of the system's statistical models.
ELFI: Engine for Likelihood-Free Inference
Jul 05, 2018

Engine for Likelihood-Free Inference (ELFI) is a Python software library for performing likelihood-free inference (LFI). ELFI provides a convenient syntax for arranging components in LFI, such as priors, simulators, summaries or distances, to a network called ELFI graph. The components can be implemented in a wide variety of languages. The stand-alone ELFI graph can be used with any of the available inference methods without modifications. A central method implemented in ELFI is Bayesian Optimization for Likelihood-Free Inference (BOLFI), which has recently been shown to accelerate likelihood-free inference up to several orders of magnitude by surrogate-modelling the distance. ELFI also has an inbuilt support for output data storing for reuse and analysis, and supports parallelization of computation from multiple cores up to a cluster environment. ELFI is designed to be extensible and provides interfaces for widening its functionality. This makes the adding of new inference methods to ELFI straightforward and automatically compatible with the inbuilt features.
Inverse Reinforcement Learning from Summary Data
Jun 17, 2018

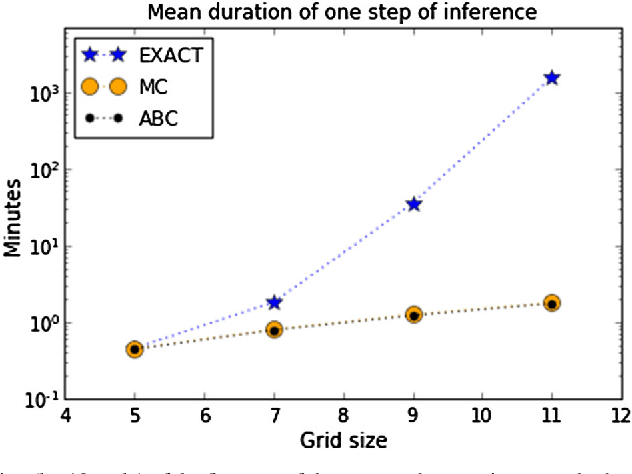
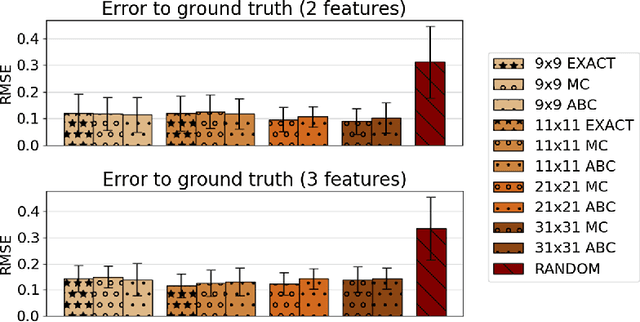
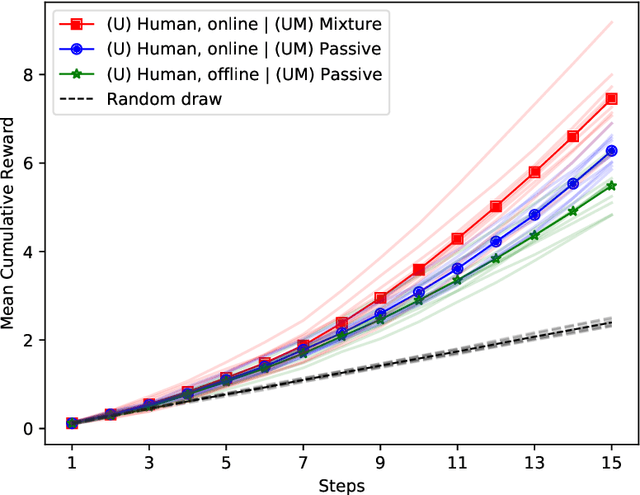
Inverse reinforcement learning (IRL) aims to explain observed strategic behavior by fitting reinforcement learning models to behavioral data. However, traditional IRL methods are only applicable when the observations are in the form of state-action paths. This assumption may not hold in many real-world modeling settings, where only partial or summarized observations are available. In general, we may assume that there is a summarizing function $\sigma$, which acts as a filter between us and the true state-action paths that constitute the demonstration. Some initial approaches to extending IRL to such situations have been presented, but with very specific assumptions about the structure of $\sigma$, such as that only certain state observations are missing. This paper instead focuses on the most general case of the problem, where no assumptions are made about the summarizing function, except that it can be evaluated. We demonstrate that inference is still possible. The paper presents exact and approximate inference algorithms that allow full posterior inference, which is particularly important for assessing parameter uncertainty in this challenging inference situation. Empirical scalability is demonstrated to reasonably sized problems, and practical applicability is demonstrated by estimating the posterior for a cognitive science RL model based on an observed user's task completion time only.
Bayesian Metabolic Flux Analysis reveals intracellular flux couplings
Apr 18, 2018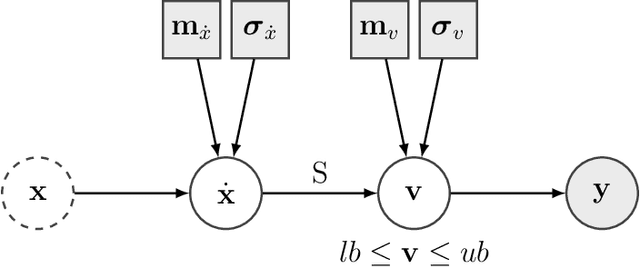
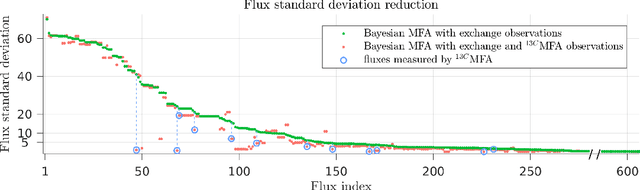
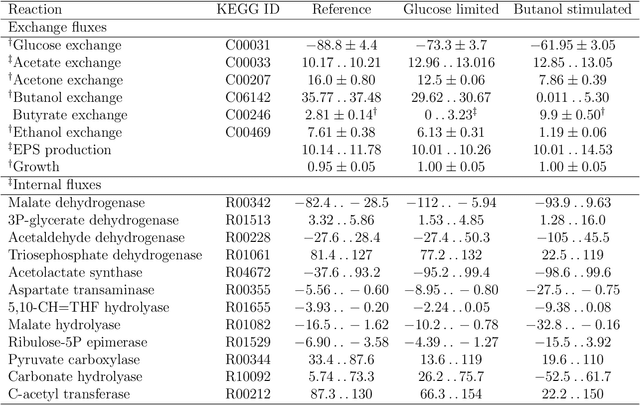
Metabolic flux balance analyses are a standard tool in analysing metabolic reaction rates compatible with measurements, steady-state and the metabolic reaction network stoichiometry. Flux analysis methods commonly place unrealistic assumptions on fluxes due to the convenience of formulating the problem as a linear programming model, and most methods ignore the notable uncertainty in flux estimates. We introduce a novel paradigm of Bayesian metabolic flux analysis that models the reactions of the whole genome-scale cellular system in probabilistic terms, and can infer the full flux vector distribution of genome-scale metabolic systems based on exchange and intracellular (e.g. 13C) flux measurements, steady-state assumptions, and target function assumptions. The Bayesian model couples all fluxes jointly together in a simple truncated multivariate posterior distribution, which reveals informative flux couplings. Our model is a plug-in replacement to conventional metabolic balance methods, such as flux balance analysis (FBA). Our experiments indicate that we can characterise the genome-scale flux covariances, reveal flux couplings, and determine more intracellular unobserved fluxes in C. acetobutylicum from 13C data than flux variability analysis. The COBRA compatible software is available at github.com/markusheinonen/bamfa
Variational zero-inflated Gaussian processes with sparse kernels
Mar 13, 2018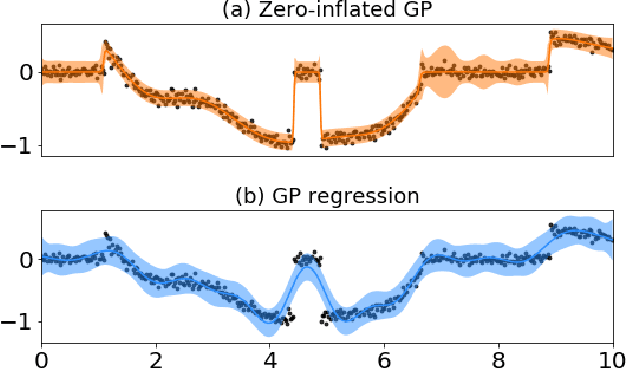
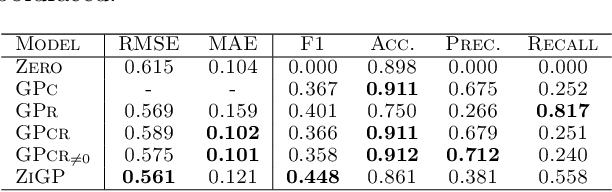
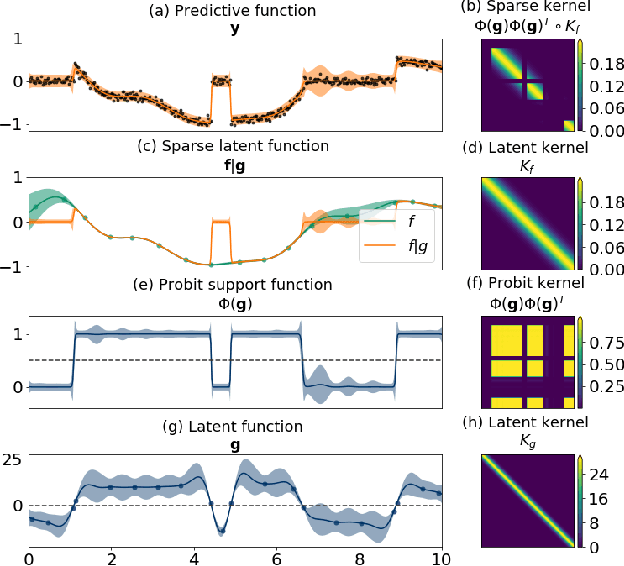
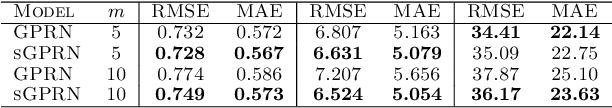
Zero-inflated datasets, which have an excess of zero outputs, are commonly encountered in problems such as climate or rare event modelling. Conventional machine learning approaches tend to overestimate the non-zeros leading to poor performance. We propose a novel model family of zero-inflated Gaussian processes (ZiGP) for such zero-inflated datasets, produced by sparse kernels through learning a latent probit Gaussian process that can zero out kernel rows and columns whenever the signal is absent. The ZiGPs are particularly useful for making the powerful Gaussian process networks more interpretable. We introduce sparse GP networks where variable-order latent modelling is achieved through sparse mixing signals. We derive the non-trivial stochastic variational inference tractably for scalable learning of the sparse kernels in both models. The novel output-sparse approach improves both prediction of zero-inflated data and interpretability of latent mixing models.
User Modelling for Avoiding Overfitting in Interactive Knowledge Elicitation for Prediction
Mar 09, 2018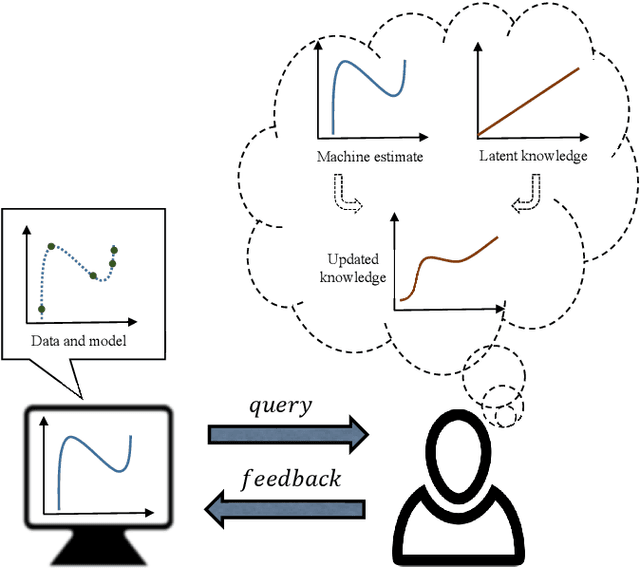

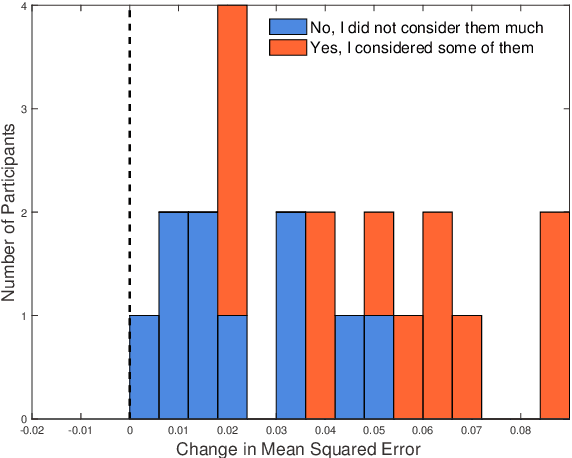
In human-in-the-loop machine learning, the user provides information beyond that in the training data. Many algorithms and user interfaces have been designed to optimize and facilitate this human--machine interaction; however, fewer studies have addressed the potential defects the designs can cause. Effective interaction often requires exposing the user to the training data or its statistics. The design of the system is then critical, as this can lead to double use of data and overfitting, if the user reinforces noisy patterns in the data. We propose a user modelling methodology, by assuming simple rational behaviour, to correct the problem. We show, in a user study with 48 participants, that the method improves predictive performance in a sparse linear regression sentiment analysis task, where graded user knowledge on feature relevance is elicited. We believe that the key idea of inferring user knowledge with probabilistic user models has general applicability in guarding against overfitting and improving interactive machine learning.
Distributed Bayesian Matrix Factorization with Limited Communication
Feb 13, 2018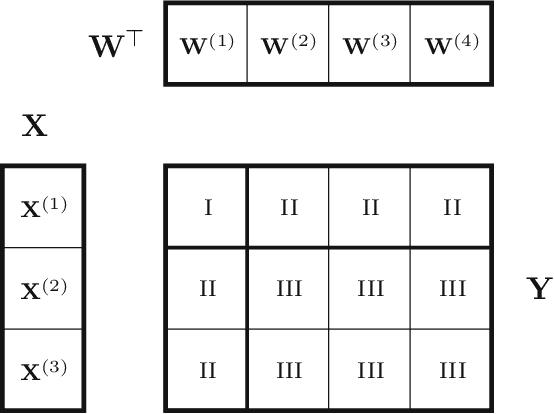
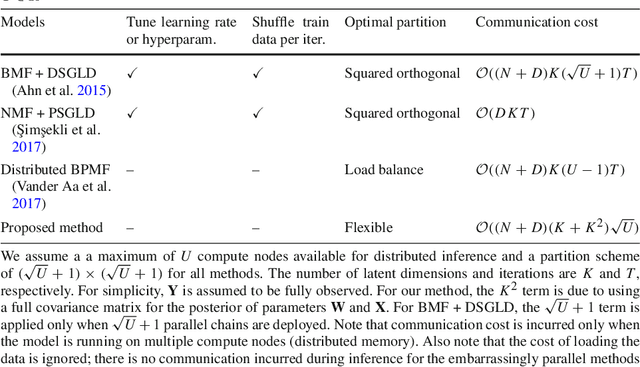
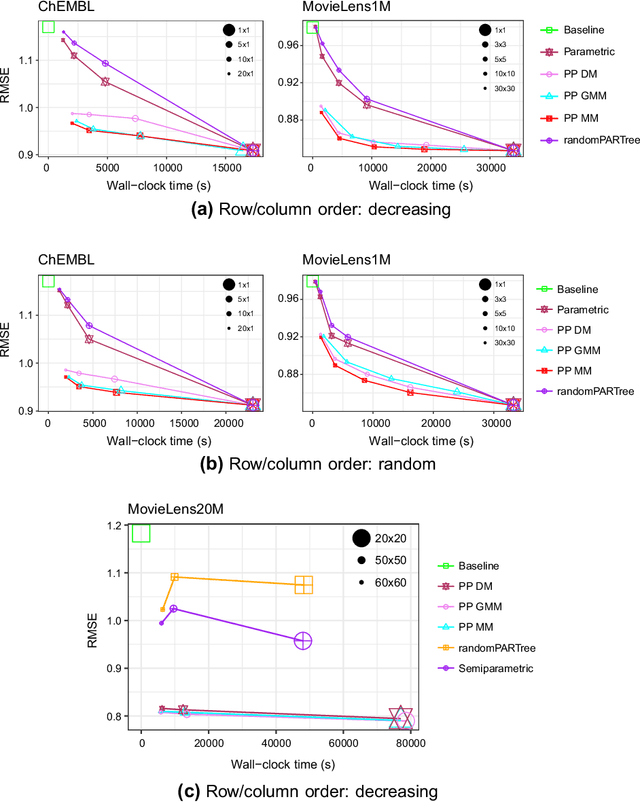
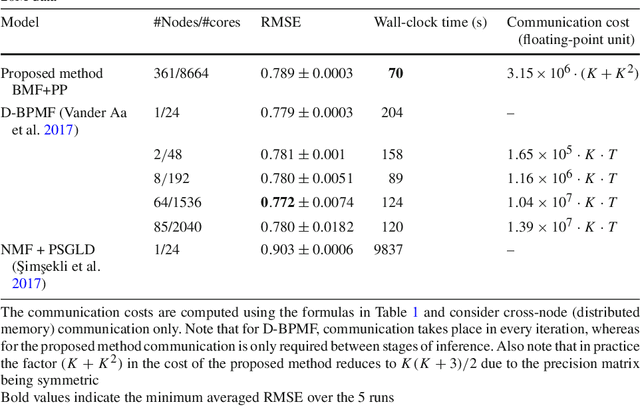
Bayesian matrix factorization (BMF) is a powerful tool for producing low-rank representations of matrices and for predicting missing values and their confidence intervals. Scaling up the posterior inference for massive-scale matrices is challenging and requires distributing both data and computation over many workers, making communication the main computational bottleneck. Embarrassingly parallel inference would remove the communication needed, by using completely independent computations on different data subsets, but suffers from the inherent unidentifiability of BMF solutions. We introduce a hierarchical decomposition of the joint posterior distribution, which couples the subset inferences, allowing for embarrassingly parallel computations in a sequence of at most three stages. Using an efficient approximate implementation, we show empirically on both real and simulated data that our distributed approach is able to achieve a speed-up of almost an order of magnitude, with a negligible effect on predictive accuracy.
A Mutually-Dependent Hadamard Kernel for Modelling Latent Variable Couplings
Oct 03, 2017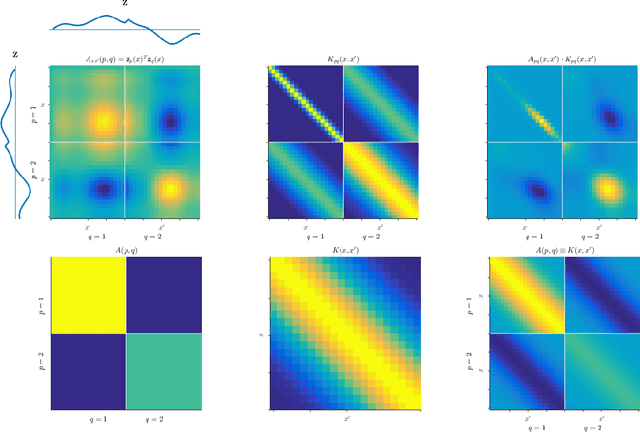

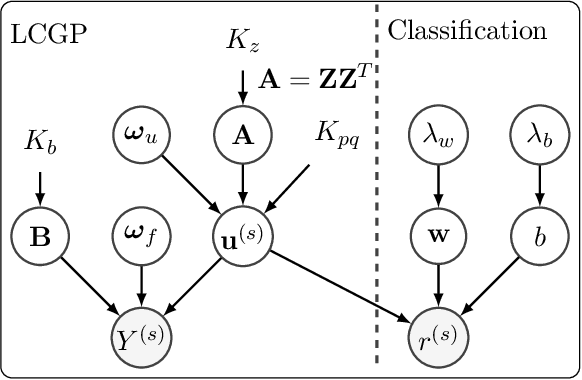
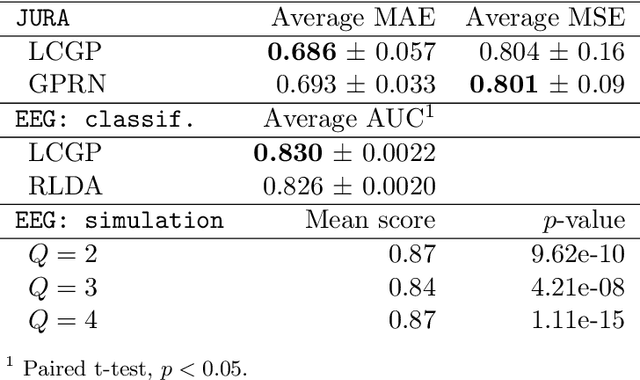
We introduce a novel kernel that models input-dependent couplings across multiple latent processes. The pairwise joint kernel measures covariance along inputs and across different latent signals in a mutually-dependent fashion. A latent correlation Gaussian process (LCGP) model combines these non-stationary latent components into multiple outputs by an input-dependent mixing matrix. Probit classification and support for multiple observation sets are derived by Variational Bayesian inference. Results on several datasets indicate that the LCGP model can recover the correlations between latent signals while simultaneously achieving state-of-the-art performance. We highlight the latent covariances with an EEG classification dataset where latent brain processes and their couplings simultaneously emerge from the model.