 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSE(3) diffusion model with application to protein backbone generation
Feb 11, 2023



The design of novel protein structures remains a challenge in protein engineering for applications across biomedicine and chemistry. In this line of work, a diffusion model over rigid bodies in 3D (referred to as frames) has shown success in generating novel, functional protein backbones that have not been observed in nature. However, there exists no principled methodological framework for diffusion on SE(3), the space of orientation preserving rigid motions in R3, that operates on frames and confers the group invariance. We address these shortcomings by developing theoretical foundations of SE(3) invariant diffusion models on multiple frames followed by a novel framework, FrameDiff, for learning the SE(3) equivariant score over multiple frames. We apply FrameDiff on monomer backbone generation and find it can generate designable monomers up to 500 amino acids without relying on a pretrained protein structure prediction network that has been integral to previous methods. We find our samples are capable of generalizing beyond any known protein structure.
Efficiently Controlling Multiple Risks with Pareto Testing
Oct 14, 2022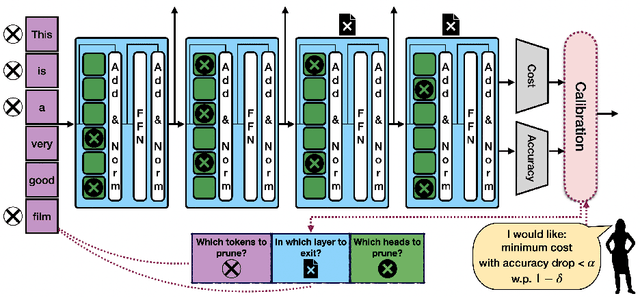
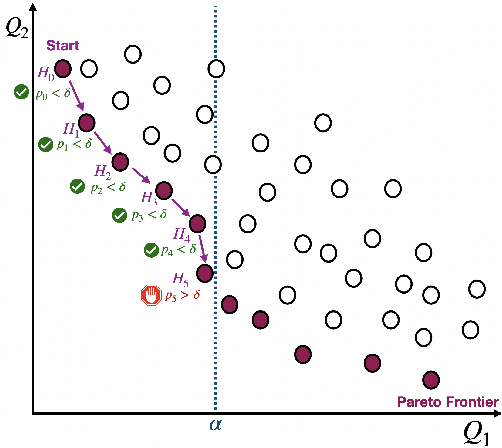
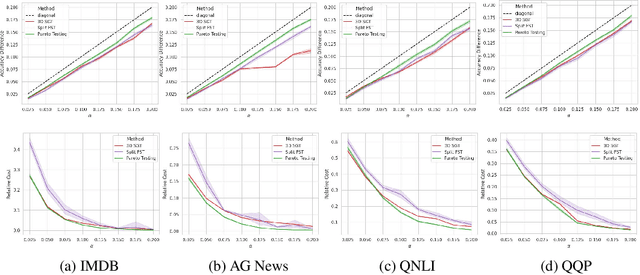
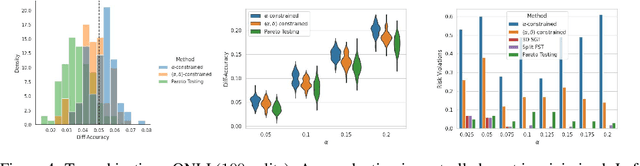
Machine learning applications frequently come with multiple diverse objectives and constraints that can change over time. Accordingly, trained models can be tuned with sets of hyper-parameters that affect their predictive behavior (e.g., their run-time efficiency versus error rate). As the number of constraints and hyper-parameter dimensions grow, naively selected settings may lead to sub-optimal and/or unreliable results. We develop an efficient method for calibrating models such that their predictions provably satisfy multiple explicit and simultaneous statistical guarantees (e.g., upper-bounded error rates), while also optimizing any number of additional, unconstrained objectives (e.g., total run-time cost). Building on recent results in distribution-free, finite-sample risk control for general losses, we propose Pareto Testing: a two-stage process which combines multi-objective optimization with multiple hypothesis testing. The optimization stage constructs a set of promising combinations on the Pareto frontier. We then apply statistical testing to this frontier only to identify configurations that have (i) high utility with respect to our objectives, and (ii) guaranteed risk levels with respect to our constraints, with specifiable high probability. We demonstrate the effectiveness of our approach to reliably accelerate the execution of large-scale Transformer models in natural language processing (NLP) applications. In particular, we show how Pareto Testing can be used to dynamically configure multiple inter-dependent model attributes -- including the number of layers computed before exiting, number of attention heads pruned, or number of text tokens considered -- to simultaneously control and optimize various accuracy and cost metrics.
DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
Oct 04, 2022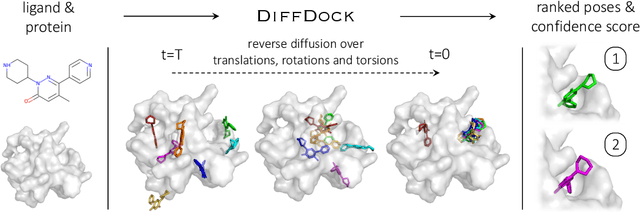
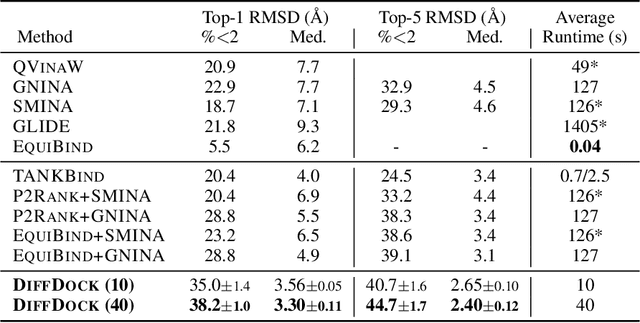
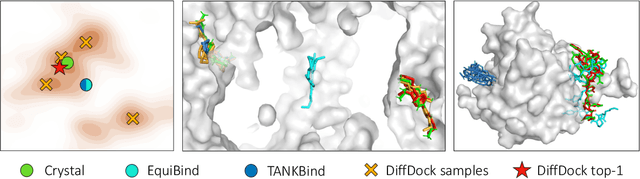

Predicting the binding structure of a small molecule ligand to a protein -- a task known as molecular docking -- is critical to drug design. Recent deep learning methods that treat docking as a regression problem have decreased runtime compared to traditional search-based methods but have yet to offer substantial improvements in accuracy. We instead frame molecular docking as a generative modeling problem and develop DiffDock, a diffusion generative model over the non-Euclidean manifold of ligand poses. To do so, we map this manifold to the product space of the degrees of freedom (translational, rotational, and torsional) involved in docking and develop an efficient diffusion process on this space. Empirically, DiffDock obtains a 38% top-1 success rate (RMSD<2A) on PDBBind, significantly outperforming the previous state-of-the-art of traditional docking (23%) and deep learning (20%) methods. Moreover, DiffDock has fast inference times and provides confidence estimates with high selective accuracy.
Calibrated Selective Classification
Aug 25, 2022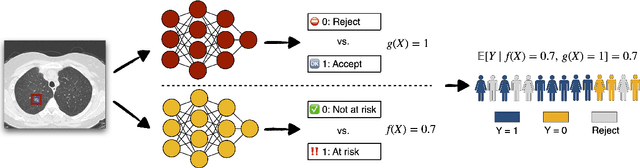
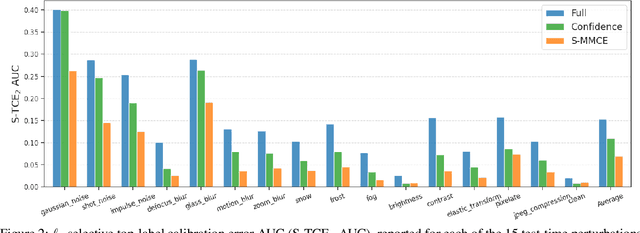
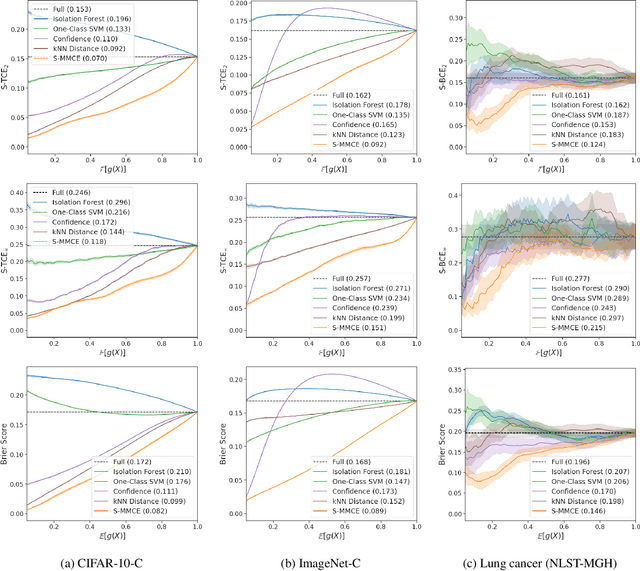
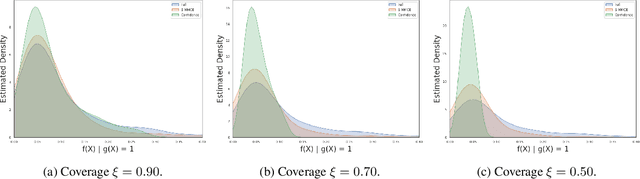
Selective classification allows models to abstain from making predictions (e.g., say "I don't know") when in doubt in order to obtain better effective accuracy. While typical selective models can be effective at producing more accurate predictions on average, they may still allow for wrong predictions that have high confidence, or skip correct predictions that have low confidence. Providing calibrated uncertainty estimates alongside predictions -- probabilities that correspond to true frequencies -- can be as important as having predictions that are simply accurate on average. However, uncertainty estimates can be unreliable for certain inputs. In this paper, we develop a new approach to selective classification in which we propose a method for rejecting examples with "uncertain" uncertainties. By doing so, we aim to make predictions with {well-calibrated} uncertainty estimates over the distribution of accepted examples, a property we call selective calibration. We present a framework for learning selectively calibrated models, where a separate selector network is trained to improve the selective calibration error of a given base model. In particular, our work focuses on achieving robust calibration, where the model is intentionally designed to be tested on out-of-domain data. We achieve this through a training strategy inspired by distributionally robust optimization, in which we apply simulated input perturbations to the known, in-domain training data. We demonstrate the empirical effectiveness of our approach on multiple image classification and lung cancer risk assessment tasks.
Antibody-Antigen Docking and Design via Hierarchical Equivariant Refinement
Jul 14, 2022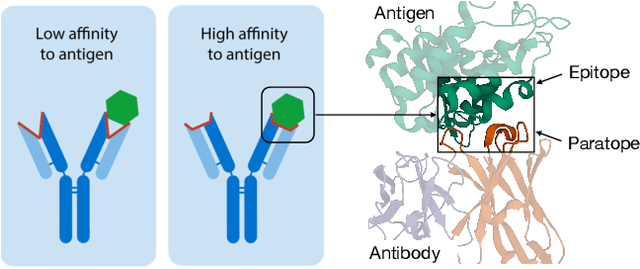
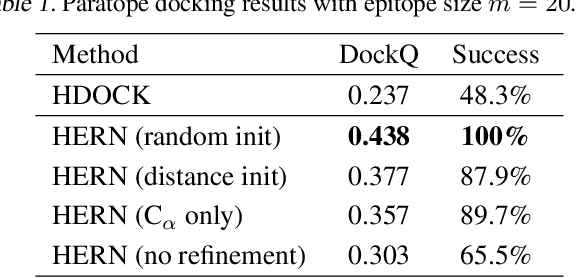
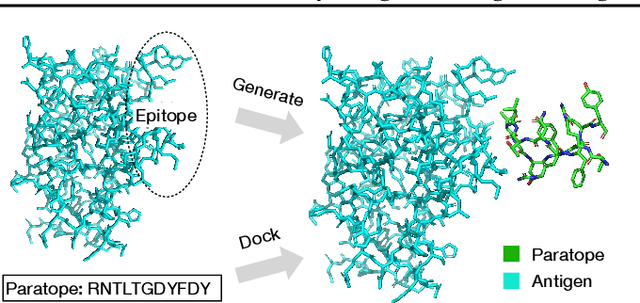
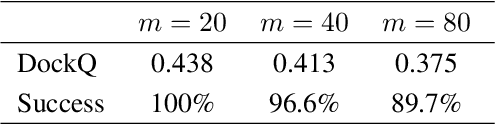
Computational antibody design seeks to automatically create an antibody that binds to an antigen. The binding affinity is governed by the 3D binding interface where antibody residues (paratope) closely interact with antigen residues (epitope). Thus, predicting 3D paratope-epitope complex (docking) is the key to finding the best paratope. In this paper, we propose a new model called Hierarchical Equivariant Refinement Network (HERN) for paratope docking and design. During docking, HERN employs a hierarchical message passing network to predict atomic forces and use them to refine a binding complex in an iterative, equivariant manner. During generation, its autoregressive decoder progressively docks generated paratopes and builds a geometric representation of the binding interface to guide the next residue choice. Our results show that HERN significantly outperforms prior state-of-the-art on paratope docking and design benchmarks.
Diffusion probabilistic modeling of protein backbones in 3D for the motif-scaffolding problem
Jun 08, 2022



Construction of a scaffold structure that supports a desired motif, conferring protein function, shows promise for the design of vaccines and enzymes. But a general solution to this motif-scaffolding problem remains open. Current machine-learning techniques for scaffold design are either limited to unrealistically small scaffolds (up to length 20) or struggle to produce multiple diverse scaffolds. We propose to learn a distribution over diverse and longer protein backbone structures via an E(3)-equivariant graph neural network. We develop SMCDiff to efficiently sample scaffolds from this distribution conditioned on a given motif; our algorithm is the first to theoretically guarantee conditional samples from a diffusion model in the large-compute limit. We evaluate our designed backbones by how well they align with AlphaFold2-predicted structures. We show that our method can (1) sample scaffolds up to 80 residues and (2) achieve structurally diverse scaffolds for a fixed motif.
Torsional Diffusion for Molecular Conformer Generation
Jun 01, 2022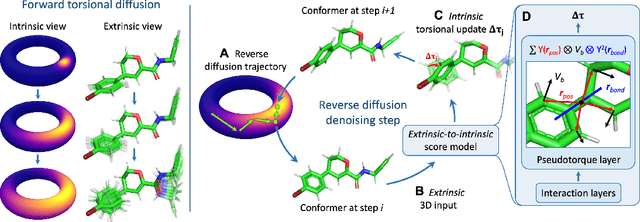

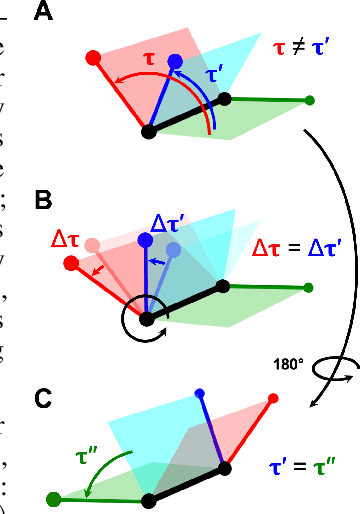
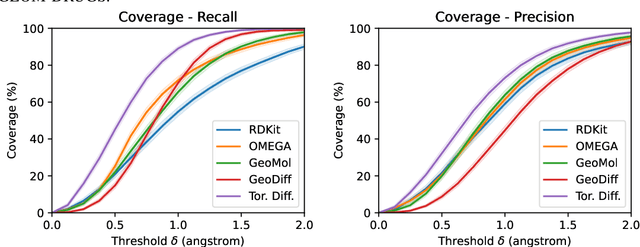
Molecular conformer generation is a fundamental task in computational chemistry. Several machine learning approaches have been developed, but none have outperformed state-of-the-art cheminformatics methods. We propose torsional diffusion, a novel diffusion framework that operates on the space of torsion angles via a diffusion process on the hypertorus and an extrinsic-to-intrinsic score model. On a standard benchmark of drug-like molecules, torsional diffusion generates superior conformer ensembles compared to machine learning and cheminformatics methods in terms of both RMSD and chemical properties, and is orders of magnitude faster than previous diffusion-based models. Moreover, our model provides exact likelihoods, which we employ to build the first generalizable Boltzmann generator. Code is available at https://github.com/gcorso/torsional-diffusion.
Robust Molecular Image Recognition: A Graph Generation Approach
May 28, 2022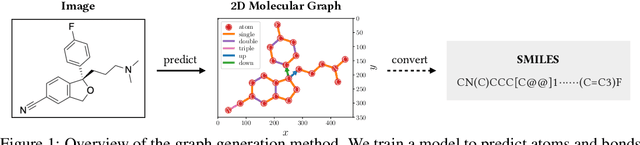
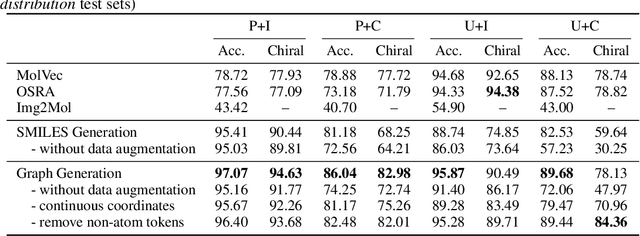
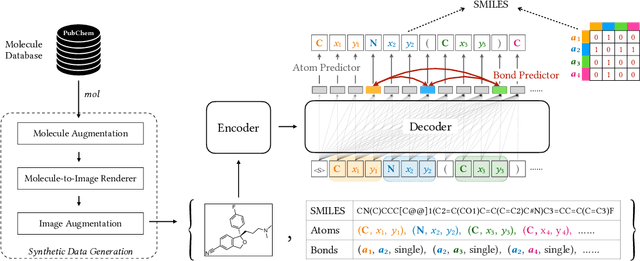
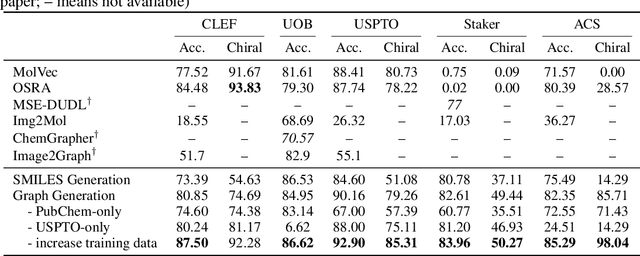
Molecular image recognition is a fundamental task in information extraction from chemistry literature. Previous data-driven models formulate it as an image-to-sequence task, to generate a sequential representation of the molecule (e.g. SMILES string) from its graphical representation. Although they perform adequately on certain benchmarks, these models are not robust in real-world situations, where molecular images differ in style, quality, and chemical patterns. In this paper, we propose a novel graph generation approach that explicitly predicts atoms and bonds, along with their geometric layouts, to construct the molecular graph. We develop data augmentation strategies for molecules and images to increase the robustness of our model against domain shifts. Our model is flexible to incorporate chemistry constraints, and produces more interpretable predictions than SMILES. In experiments on both synthetic and realistic molecular images, our model significantly outperforms previous models, achieving 84-93% accuracy on five benchmarks. We also conduct human evaluation and show that our model reduces the time for a chemist to extract molecular structures from images by roughly 50%.
Learning to Split for Automatic Bias Detection
Apr 28, 2022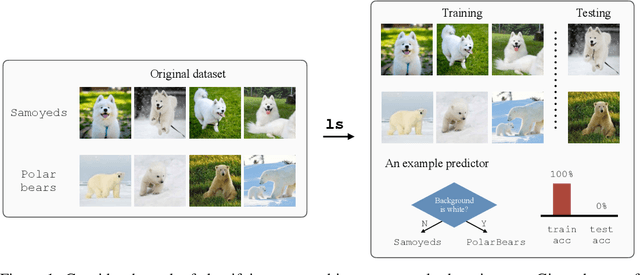

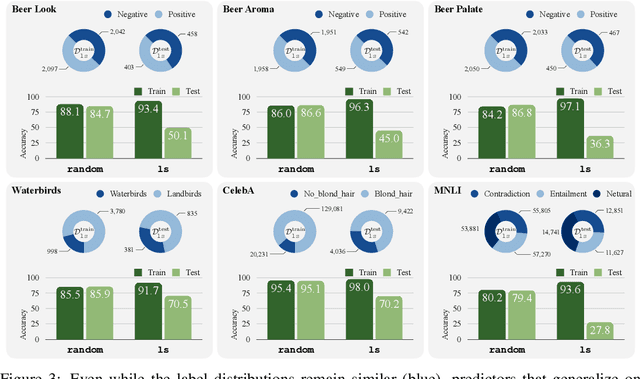
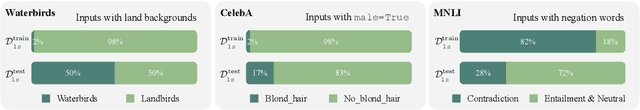
Classifiers are biased when trained on biased datasets. As a remedy, we propose Learning to Split (ls), an algorithm for automatic bias detection. Given a dataset with input-label pairs, ls learns to split this dataset so that predictors trained on the training split generalize poorly to the testing split. This performance gap provides a proxy for measuring the degree of bias in the learned features and can therefore be used to reduce biases. Identifying non-generalizable splits is challenging as we don't have any explicit annotations about how to split. In this work, we show that the prediction correctness of the testing example can be used as a source of weak supervision: generalization performance will drop if we move examples that are predicted correctly away from the testing split, leaving only those that are mispredicted. We evaluate our approach on Beer Review, Waterbirds, CelebA and MNLI. Empirical results show that ls is able to generate astonishingly challenging splits that correlate with human-identified biases. Moreover, we demonstrate that combining robust learning algorithms (such as group DRO) with splits identified by ls enables automatic de-biasing. Compared with previous state-of-the-arts, we substantially improves the worst-group performance (23.4% on average) when the source of biases is unknown during training and validation.
Conformal Prediction Sets with Limited False Positives
Feb 15, 2022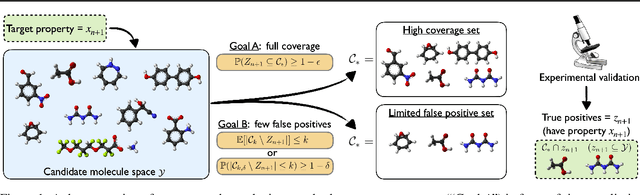
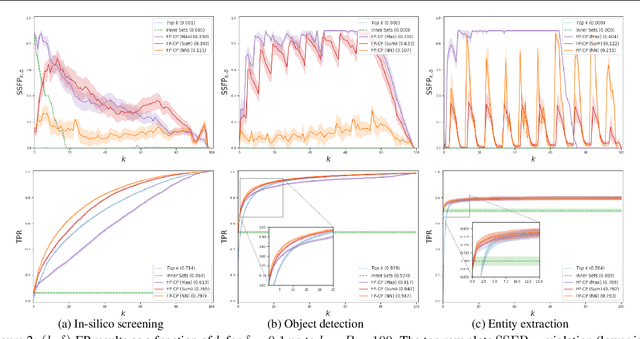
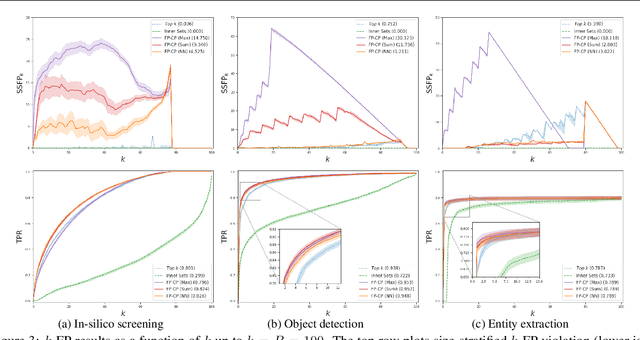
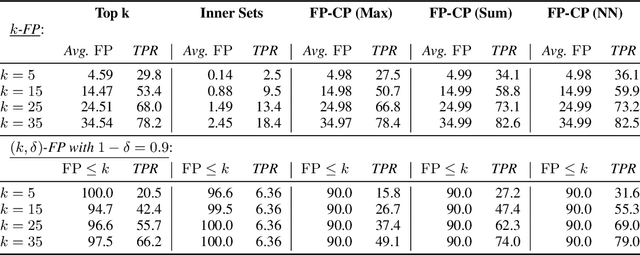
We develop a new approach to multi-label conformal prediction in which we aim to output a precise set of promising prediction candidates with a bounded number of incorrect answers. Standard conformal prediction provides the ability to adapt to model uncertainty by constructing a calibrated candidate set in place of a single prediction, with guarantees that the set contains the correct answer with high probability. In order to obey this coverage property, however, conformal sets can become inundated with noisy candidates -- which can render them unhelpful in practice. This is particularly relevant to practical applications where there is a limited budget, and the cost (monetary or otherwise) associated with false positives is non-negligible. We propose to trade coverage for a notion of precision by enforcing that the presence of incorrect candidates in the predicted conformal sets (i.e., the total number of false positives) is bounded according to a user-specified tolerance. Subject to this constraint, our algorithm then optimizes for a generalized notion of set coverage (i.e., the true positive rate) that allows for any number of true answers for a given query (including zero). We demonstrate the effectiveness of this approach across a number of classification tasks in natural language processing, computer vision, and computational chemistry.