 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHiFusion: Hierarchical Intra-Spot Alignment and Regional Context Fusion for Spatial Gene Expression Prediction from Histopathology
Nov 19, 2025Spatial transcriptomics (ST) bridges gene expression and tissue morphology but faces clinical adoption barriers due to technical complexity and prohibitive costs. While computational methods predict gene expression from H&E-stained whole-slide images (WSIs), existing approaches often fail to capture the intricate biological heterogeneity within spots and are susceptible to morphological noise when integrating contextual information from surrounding tissue. To overcome these limitations, we propose HiFusion, a novel deep learning framework that integrates two complementary components. First, we introduce the Hierarchical Intra-Spot Modeling module that extracts fine-grained morphological representations through multi-resolution sub-patch decomposition, guided by a feature alignment loss to ensure semantic consistency across scales. Concurrently, we present the Context-aware Cross-scale Fusion module, which employs cross-attention to selectively incorporate biologically relevant regional context, thereby enhancing representational capacity. This architecture enables comprehensive modeling of both cellular-level features and tissue microenvironmental cues, which are essential for accurate gene expression prediction. Extensive experiments on two benchmark ST datasets demonstrate that HiFusion achieves state-of-the-art performance across both 2D slide-wise cross-validation and more challenging 3D sample-specific scenarios. These results underscore HiFusion's potential as a robust, accurate, and scalable solution for ST inference from routine histopathology.
Novel Clinical-Grade Prostate Cancer Detection and Grading Model: Development and Prospective Validation Using Real World Data, with Performance Assessment on IHC Requested Cases
Oct 31, 2024



Artificial intelligence may assist healthcare systems in meeting increasing demand for pathology services while maintaining diagnostic quality and reducing turnaround time and costs. We aimed to investigate the performance of an institutionally developed system for prostate cancer detection, grading, and workflow optimization and to contrast this with commercial alternatives. From August 2021 to March 2023, we scanned 21,396 slides from 1,147 patients with positive biopsies. We developed models for cancer detection, grading, and screening of equivocal cases for IHC ordering. We compared a task-specific model trained using the PANDA dataset of prostate cancer biopsies with one built using features extracted by the general-purpose histology foundation model, UNI and compare their performance in an unfiltered prospectively collected dataset that reflects our patient population (1737 slides,95 patients). We evaluated the contributions of a bespoke model designed to improve sensitivity in detecting small cancer foci and scoring of broader patterns observed at lower resolution. We found high concordance between the developed systems and pathologist reference in detection (AUC 98.5, sensitivity 95.0, and specificity 97.8), ISUP grading (quadratic Cohen's kappa 0.869), grade group 3 or higher (AUC 97.5, sensitivity 94.9, specificity 96.6) and comparable to published data from commercial systems. Screening could reduce IHC ordering for equivocal cases by 44.5% with an overall error rate of 1.8% (1.4% false positive, 0.4% false negative rates). Institutions like academic medical centers that have high scanning volumes and report abstraction capabilities can develop accurate computational pathology models for internal use. These models have the potential to aid in quality control role and to improve workflow in the pathology lab to help meet future challenges in prostate cancer diagnosis.
HistomicsML2.0: Fast interactive machine learning for whole slide imaging data
Jan 30, 2020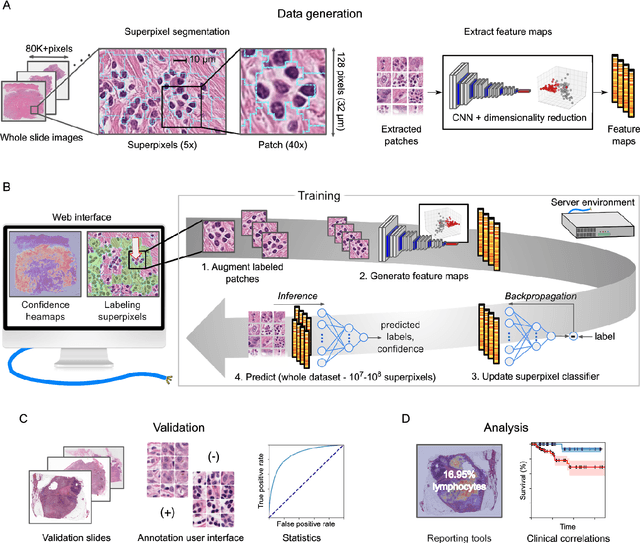
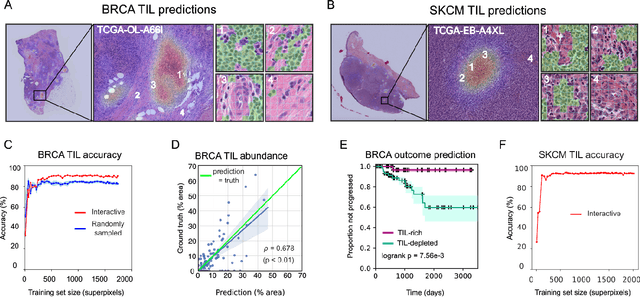
Extracting quantitative phenotypic information from whole-slide images presents significant challenges for investigators who are not experienced in developing image analysis algorithms. We present new software that enables rapid learn-by-example training of machine learning classifiers for detection of histologic patterns in whole-slide imaging datasets. HistomicsML2.0 uses convolutional networks to be readily adaptable to a variety of applications, provides a web-based user interface, and is available as a software container to simplify deployment.