 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSymb-xMIL: Symbolic Explanations for Multiple Instance Learning in Digital Pathology
Jun 04, 2026Explanations of multiple instance learning (MIL) models are widely used for validation and discovery in digital histopathology. Existing methods primarily rely on heatmaps that highlight influential regions but do not explain how evidence from different tissue regions is combined to produce a prediction. This limits interpretability, especially when decisions depend on interactions between tissue features. We introduce Symbolic explainable MIL (Symb-xMIL), a post-hoc explanation framework that quantifies how a MIL model's behavior aligns with human-readable decision rules, expressed as logical relationships (e.g., AND, OR, NOT) between input features. These alignment scores reveal semantic patterns underlying the model's predictions. We evaluate Symb-xMIL on synthetic and real-world histopathology datasets. On synthetic MIL data, Symb-xMIL reliably recovers ground-truth logical rules. In a clinical tumor detection task, the best-aligned rules uncover heterogeneous decision patterns and expose hidden model errors. On an HPV-prediction task on TCGA-HNSCC, a cohort of head and neck cancer, our framework refines patient survival stratification beyond HPV status with potential clinical relevance. Overall, Symb-xMIL extends MIL explainability beyond visual attribution toward structured, rule-based reasoning, enabling more transparent and semantically grounded interpretation of model predictions.
In-Context Multiple Instance Learning
Jun 04, 2026Multiple Instance Learning (MIL) addresses problems where supervision is available at the level of bags of instances and has been successfully applied in fields ranging from computational pathology to satellite imagery. Nevertheless, existing algorithms struggle in the low-label regime that characterizes many real-world applications. Flexible models overfit and rigid ones fail to adapt to the task at hand. We show that pretraining an in-context learner with a Perceiver-style architecture on synthetic data yields a model that can solve new tasks from a handful of labeled bags. At inference time, classification happens in a single forward pass and requires no gradient updates. We propose and investigate different synthetic data generators for bag-structured data and find that they capture complementary inductive biases. A model pretrained on a mixture of these generators inherits their per-task strengths and achieves the best average performance across twelve MIL benchmarks, outperforming supervised baselines that require task-specific training.
Beyond Attention Heatmaps: How to Get Better Explanations for Multiple Instance Learning Models in Histopathology
Mar 09, 2026Multiple instance learning (MIL) has enabled substantial progress in computational histopathology, where a large amount of patches from gigapixel whole slide images are aggregated into slide-level predictions. Heatmaps are widely used to validate MIL models and to discover tissue biomarkers. Yet, the validity of these heatmaps has barely been investigated. In this work, we introduce a general framework for evaluating the quality of MIL heatmaps without requiring additional labels. We conduct a large-scale benchmark experiment to assess six explanation methods across histopathology task types (classification, regression, survival), MIL model architectures (Attention-, Transformer-, Mamba-based), and patch encoder backbones (UNI2, Virchow2). Our results show that explanation quality mostly depends on MIL model architecture and task type, with perturbation ("Single"), layer-wise relevance propagation (LRP), and integrated gradients (IG) consistently outperforming attention-based and gradient-based saliency heatmaps, which often fail to reflect model decision mechanisms. We further demonstrate the advanced capabilities of the best-performing explanation methods: (i) We provide a proof-of-concept that MIL heatmaps of a bulk gene expression prediction model can be correlated with spatial transcriptomics for biological validation, and (ii) showcase the discovery of distinct model strategies for predicting human papillomavirus (HPV) infection from head and neck cancer slides. Our work highlights the importance of validating MIL heatmaps and establishes that improved explainability can enable more reliable model validation and yield biological insights, making a case for a broader adoption of explainable AI in digital pathology. Our code is provided in a public GitHub repository: https://github.com/bifold-pathomics/xMIL/tree/xmil-journal
Mind the Gap: Continuous Magnification Sampling for Pathology Foundation Models
Jan 05, 2026In histopathology, pathologists examine both tissue architecture at low magnification and fine-grained morphology at high magnification. Yet, the performance of pathology foundation models across magnifications and the effect of magnification sampling during training remain poorly understood. We model magnification sampling as a multi-source domain adaptation problem and develop a simple theoretical framework that reveals systematic trade-offs between sampling strategies. We show that the widely used discrete uniform sampling of magnifications (0.25, 0.5, 1.0, 2.0 mpp) leads to degradation at intermediate magnifications. We introduce continuous magnification sampling, which removes gaps in magnification coverage while preserving performance at standard scales. Further, we derive sampling distributions that optimize representation quality across magnification scales. To evaluate these strategies, we introduce two new benchmarks (TCGA-MS, BRACS-MS) with appropriate metrics. Our experiments show that continuous sampling substantially improves over discrete sampling at intermediate magnifications, with gains of up to 4 percentage points in balanced classification accuracy, and that optimized distributions can further improve performance. Finally, we evaluate current histopathology foundation models, finding that magnification is a primary driver of performance variation across models. Our work paves the way towards future pathology foundation models that perform reliably across magnifications.
Towards Robust Foundation Models for Digital Pathology
Jul 22, 2025
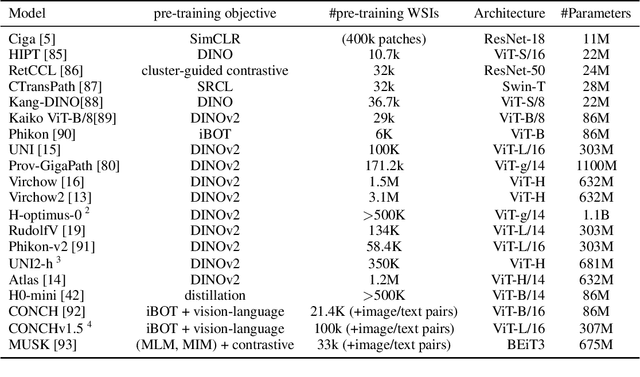
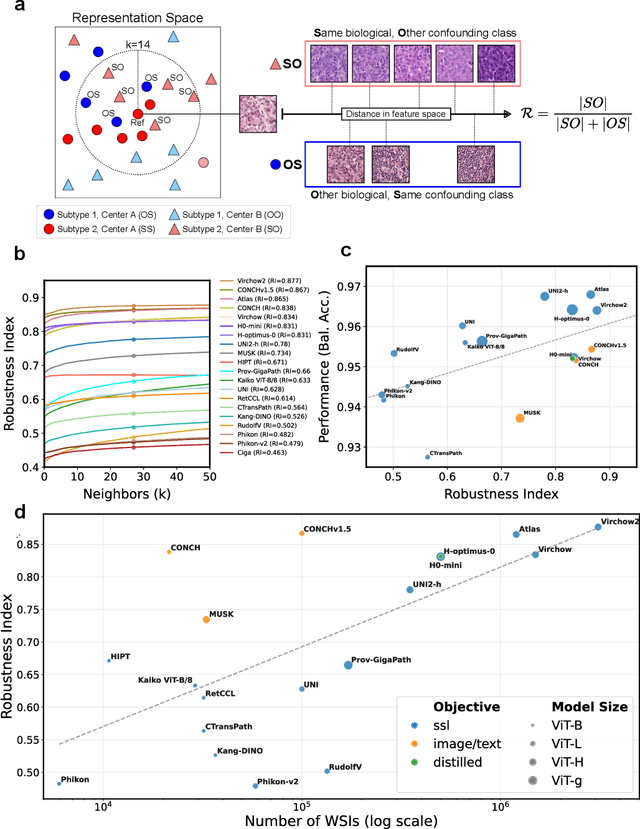

Biomedical Foundation Models (FMs) are rapidly transforming AI-enabled healthcare research and entering clinical validation. However, their susceptibility to learning non-biological technical features -- including variations in surgical/endoscopic techniques, laboratory procedures, and scanner hardware -- poses risks for clinical deployment. We present the first systematic investigation of pathology FM robustness to non-biological features. Our work (i) introduces measures to quantify FM robustness, (ii) demonstrates the consequences of limited robustness, and (iii) proposes a framework for FM robustification to mitigate these issues. Specifically, we developed PathoROB, a robustness benchmark with three novel metrics, including the robustness index, and four datasets covering 28 biological classes from 34 medical centers. Our experiments reveal robustness deficits across all 20 evaluated FMs, and substantial robustness differences between them. We found that non-robust FM representations can cause major diagnostic downstream errors and clinical blunders that prevent safe clinical adoption. Using more robust FMs and post-hoc robustification considerably reduced (but did not yet eliminate) the risk of such errors. This work establishes that robustness evaluation is essential for validating pathology FMs before clinical adoption and demonstrates that future FM development must integrate robustness as a core design principle. PathoROB provides a blueprint for assessing robustness across biomedical domains, guiding FM improvement efforts towards more robust, representative, and clinically deployable AI systems that prioritize biological information over technical artifacts.
Do Histopathological Foundation Models Eliminate Batch Effects? A Comparative Study
Nov 08, 2024



Deep learning has led to remarkable advancements in computational histopathology, e.g., in diagnostics, biomarker prediction, and outcome prognosis. Yet, the lack of annotated data and the impact of batch effects, e.g., systematic technical data differences across hospitals, hamper model robustness and generalization. Recent histopathological foundation models -- pretrained on millions to billions of images -- have been reported to improve generalization performances on various downstream tasks. However, it has not been systematically assessed whether they fully eliminate batch effects. In this study, we empirically show that the feature embeddings of the foundation models still contain distinct hospital signatures that can lead to biased predictions and misclassifications. We further find that the signatures are not removed by stain normalization methods, dominate distances in feature space, and are evident across various principal components. Our work provides a novel perspective on the evaluation of medical foundation models, paving the way for more robust pretraining strategies and downstream predictors.
AI-based Anomaly Detection for Clinical-Grade Histopathological Diagnostics
Jun 21, 2024



While previous studies have demonstrated the potential of AI to diagnose diseases in imaging data, clinical implementation is still lagging behind. This is partly because AI models require training with large numbers of examples only available for common diseases. In clinical reality, however, only few diseases are common, whereas the majority of diseases are less frequent (long-tail distribution). Current AI models overlook or misclassify these diseases. We propose a deep anomaly detection approach that only requires training data from common diseases to detect also all less frequent diseases. We collected two large real-world datasets of gastrointestinal biopsies, which are prototypical of the problem. Herein, the ten most common findings account for approximately 90% of cases, whereas the remaining 10% contained 56 disease entities, including many cancers. 17 million histological images from 5,423 cases were used for training and evaluation. Without any specific training for the diseases, our best-performing model reliably detected a broad spectrum of infrequent ("anomalous") pathologies with 95.0% (stomach) and 91.0% (colon) AUROC and generalized across scanners and hospitals. By design, the proposed anomaly detection can be expected to detect any pathological alteration in the diagnostic tail of gastrointestinal biopsies, including rare primary or metastatic cancers. This study establishes the first effective clinical application of AI-based anomaly detection in histopathology that can flag anomalous cases, facilitate case prioritization, reduce missed diagnoses and enhance the general safety of AI models, thereby driving AI adoption and automation in routine diagnostics and beyond.
xMIL: Insightful Explanations for Multiple Instance Learning in Histopathology
Jun 06, 2024



Multiple instance learning (MIL) is an effective and widely used approach for weakly supervised machine learning. In histopathology, MIL models have achieved remarkable success in tasks like tumor detection, biomarker prediction, and outcome prognostication. However, MIL explanation methods are still lagging behind, as they are limited to small bag sizes or disregard instance interactions. We revisit MIL through the lens of explainable AI (XAI) and introduce xMIL, a refined framework with more general assumptions. We demonstrate how to obtain improved MIL explanations using layer-wise relevance propagation (LRP) and conduct extensive evaluation experiments on three toy settings and four real-world histopathology datasets. Our approach consistently outperforms previous explanation attempts with particularly improved faithfulness scores on challenging biomarker prediction tasks. Finally, we showcase how xMIL explanations enable pathologists to extract insights from MIL models, representing a significant advance for knowledge discovery and model debugging in digital histopathology.