 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBohrium + SciMaster: Building the Infrastructure and Ecosystem for Agentic Science at Scale
Dec 23, 2025



AI agents are emerging as a practical way to run multi-step scientific workflows that interleave reasoning with tool use and verification, pointing to a shift from isolated AI-assisted steps toward \emph{agentic science at scale}. This shift is increasingly feasible, as scientific tools and models can be invoked through stable interfaces and verified with recorded execution traces, and increasingly necessary, as AI accelerates scientific output and stresses the peer-review and publication pipeline, raising the bar for traceability and credible evaluation. However, scaling agentic science remains difficult: workflows are hard to observe and reproduce; many tools and laboratory systems are not agent-ready; execution is hard to trace and govern; and prototype AI Scientist systems are often bespoke, limiting reuse and systematic improvement from real workflow signals. We argue that scaling agentic science requires an infrastructure-and-ecosystem approach, instantiated in Bohrium+SciMaster. Bohrium acts as a managed, traceable hub for AI4S assets -- akin to a HuggingFace of AI for Science -- that turns diverse scientific data, software, compute, and laboratory systems into agent-ready capabilities. SciMaster orchestrates these capabilities into long-horizon scientific workflows, on which scientific agents can be composed and executed. Between infrastructure and orchestration, a \emph{scientific intelligence substrate} organizes reusable models, knowledge, and components into executable building blocks for workflow reasoning and action, enabling composition, auditability, and improvement through use. We demonstrate this stack with eleven representative master agents in real workflows, achieving orders-of-magnitude reductions in end-to-end scientific cycle time and generating execution-grounded signals from real workloads at multi-million scale.
Neural Network Based in Silico Simulation of Combustion Reactions
Nov 27, 2019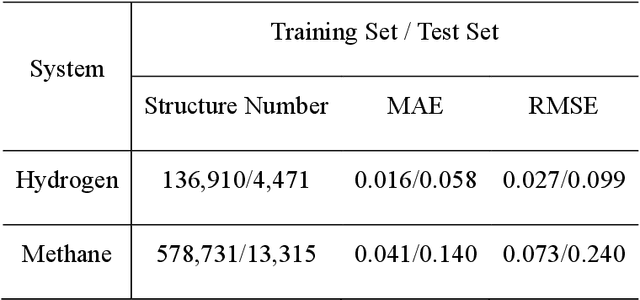
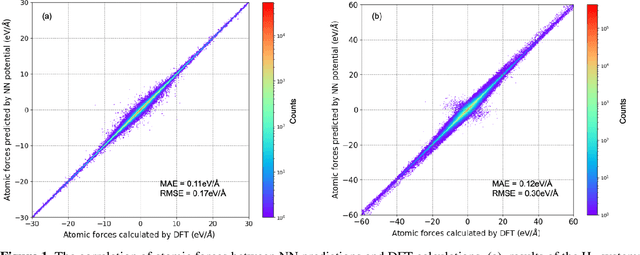
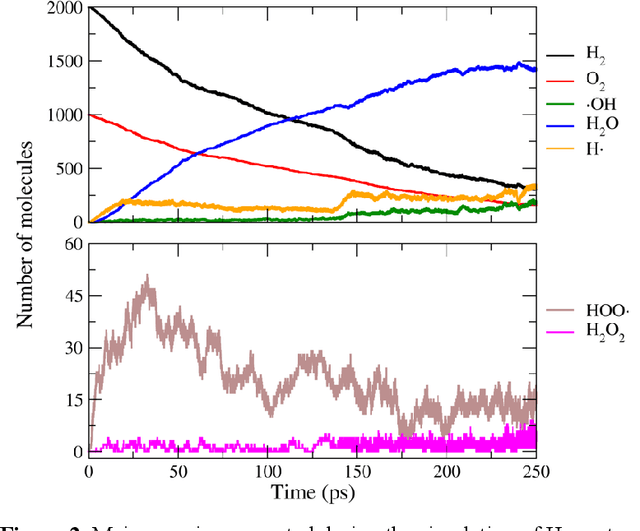
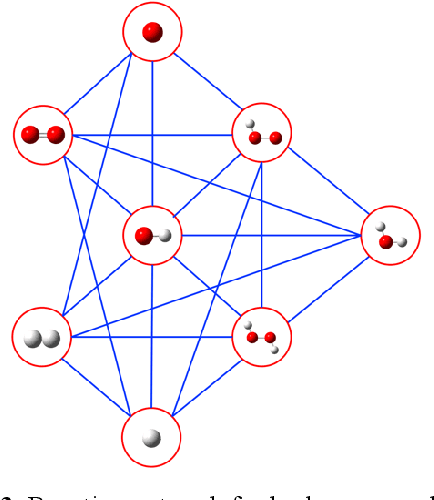
Understanding and prediction of the chemical reactions are fundamental demanding in the study of many complex chemical systems. Reactive molecular dynamics (MD) simulation has been widely used for this purpose as it can offer atomic details and can help us better interpret chemical reaction mechanisms. In this study, two reference datasets were constructed and corresponding neural network (NN) potentials were trained based on them. For given large-scale reaction systems, the NN potentials can predict the potential energy and atomic forces of DFT precision, while it is orders of magnitude faster than the conventional DFT calculation. With these two models, reactive MD simulations were performed to explore the combustion mechanisms of hydrogen and methane. Benefit from the high efficiency of the NN model, nanosecond MD trajectories for large-scale systems containing hundreds of atoms were produced and detailed combustion mechanism was obtained. Through further development, the algorithms in this study can be used to explore and discovery reaction mechanisms of many complex reaction systems, such as combustion, synthesis, and heterogeneous catalysis without any predefined reaction coordinates and elementary reaction steps.