 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLost at the End: Primacy Bias in Multimodal Retrieval-Augmented Question Answering
Jun 15, 2026Knowledge-based visual question answering (KB-VQA) lets vision-language systems answer questions that exceed their parametric knowledge by conditioning a reader on passages retrieved from a Wikipedia-scale knowledge base. In pure-text long-context LLMs, retrieved-context use follows the U-shaped "lost-in-the-middle" effect of Liu et al. (2024): information at the start and end of context is used, the middle is lost. Whether this transfers to deployed multimodal KB-VQA is open. To close this gap, we design the first controlled probe of reader-side position dependence in multimodal KB-VQA: a gold-position protocol in which only the gold passage's prompt slot varies within question. We run it on three open-source 7B/8B VLM readers and two KB-VQA benchmarks at k up to 20. The shape flips from U to primacy: gold-at-first beats gold-at-last by 16 to 26 points on every reader-by-benchmark cell, an effect we call "Lost at the End". Three targeted ablations narrow the cause: a text-only control shows the multimodal setting amplifies an already-present text-mode primacy 2.2 to 4.5 times, and image-position and distractor-shuffle ablations together pin the locus to prompt slot 0 of the instruction-tuned reader. On a frozen reader, three retrieval-side fixes (MMR, oracle reranking, rank-based reordering) all leave the gap intact (no separable improvement). Our findings indicate that recall@k is the wrong metric for deployed KB-VQA and that closing the gap requires reader-side intervention; we release our protocol as a controlled instrument for evaluating such interventions.
CellMaster: Collaborative Cell Type Annotation in Single-Cell Analysis
Feb 12, 2026Single-cell RNA-seq (scRNA-seq) enables atlas-scale profiling of complex tissues, revealing rare lineages and transient states. Yet, assigning biologically valid cell identities remains a bottleneck because markers are tissue- and state-dependent, and novel states lack references. We present CellMaster, an AI agent that mimics expert practice for zero-shot cell-type annotation. Unlike existing automated tools, CellMaster leverages LLM-encoded knowledge (e.g., GPT-4o) to perform on-the-fly annotation with interpretable rationales, without pre-training or fixed marker databases. Across 9 datasets spanning 8 tissues, CellMaster improved accuracy by 7.1% over best-performing baselines (including CellTypist and scTab) in automatic mode. With human-in-the-loop refinement, this advantage increased to 18.6%, with a 22.1% gain on subtype populations. The system demonstrates particular strength in rare and novel cell states where baselines often fail. Source code and the web application are available at \href{https://github.com/AnonymousGym/CellMaster}{https://github.com/AnonymousGym/CellMaster}.
scPilot: Large Language Model Reasoning Toward Automated Single-Cell Analysis and Discovery
Feb 12, 2026We present scPilot, the first systematic framework to practice omics-native reasoning: a large language model (LLM) converses in natural language while directly inspecting single-cell RNA-seq data and on-demand bioinformatics tools. scPilot converts core single-cell analyses, i.e., cell-type annotation, developmental-trajectory reconstruction, and transcription-factor targeting, into step-by-step reasoning problems that the model must solve, justify, and, when needed, revise with new evidence. To measure progress, we release scBench, a suite of 9 expertly curated datasets and graders that faithfully evaluate the omics-native reasoning capability of scPilot w.r.t various LLMs. Experiments with o1 show that iterative omics-native reasoning lifts average accuracy by 11% for cell-type annotation and Gemini-2.5-Pro cuts trajectory graph-edit distance by 30% versus one-shot prompting, while generating transparent reasoning traces explain marker gene ambiguity and regulatory logic. By grounding LLMs in raw omics data, scPilot enables auditable, interpretable, and diagnostically informative single-cell analyses. Code, data, and package are available at https://github.com/maitrix-org/scPilot
Fully Automated Volumetric Classification in CT Scans for Diagnosis and Analysis of Normal Pressure Hydrocephalus
Jan 25, 2019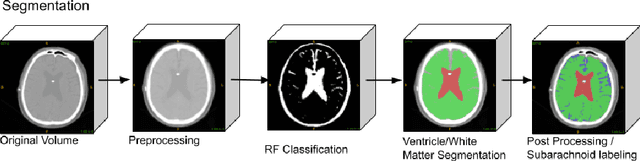
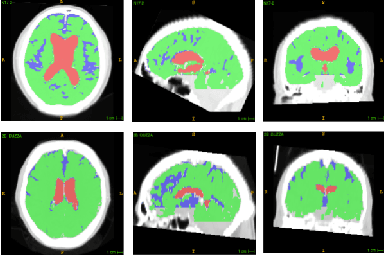
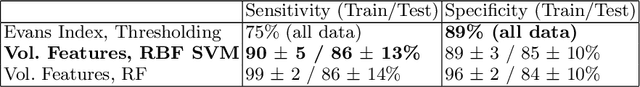
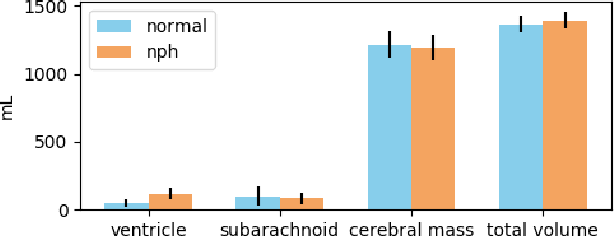
Normal Pressure Hydrocephalus (NPH) is one of the few reversible forms of dementia. Due to their low cost and versatility, Computed Tomography (CT) scans have long been used as an aid to help diagnose intracerebral anomalies such as NPH. However, because CT imaging presents 2-dimensional slices of a 3-dimensional volume, recapitulating the ventricular space in 3-dimensions to facilitate the diagnosis of NPH poses numerous challenges such as head rotation and human error. As such, no well-defined and effective protocol currently exists for the analysis of CT scan-based ventricular, white matter and subarachnoid space volumes in the setting of NPH. The Evan's ratio, an approximation of the ratio of ventricle to brain volume using only one 2D slice of the scan, has been proposed but is not robust. Instead of manually measuring a 2-dimensional proxy for the ratio of ventricle volume to brain volume, this study proposes an automated method of calculating the brain volumes for better recognition of NPH from a radiological standpoint. The method first aligns the subject CT volume to a common space through an affine transformation, then uses a random forest classifier to mask relevant tissue types. A 3D morphological segmentation method is used to partition the brain volume, which in turn is used to train machine learning methods to classify the subjects into non-NPH vs. NPH based on volumetric information.
Brain Tumor Segmentation and Tractographic Feature Extraction from Structural MR Images for Overall Survival Prediction
Oct 10, 2018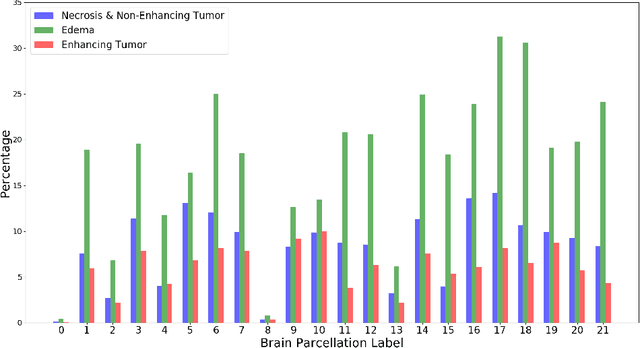
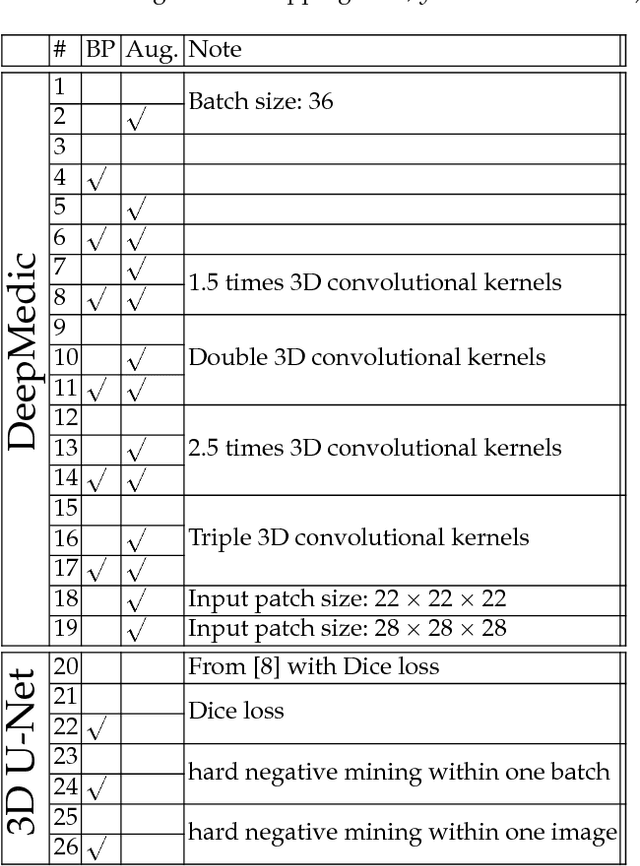
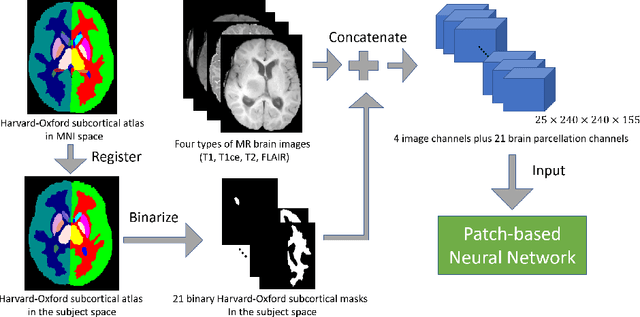
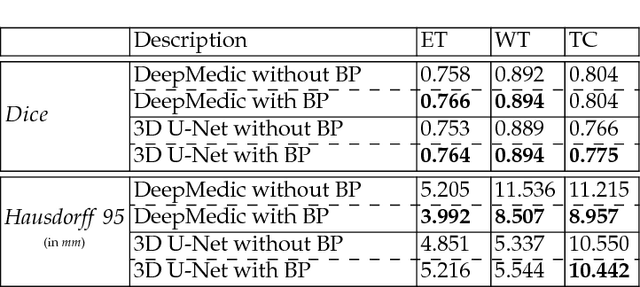
This paper introduces a novel methodology to integrate human brain connectomics and parcellation for brain tumor segmentation and survival prediction. For segmentation, we utilize an existing brain parcellation atlas in the MNI152 1mm space and map this parcellation to each individual subject data. We use deep neural network architectures together with hard negative mining to achieve the final voxel level classification. For survival prediction, we present a new method for combining features from connectomics data, brain parcellation information, and the brain tumor mask. We leverage the average connectome information from the Human Connectome Project and map each subject brain volume onto this common connectome space. From this, we compute tractographic features that describe potential neural disruptions due to the brain tumor. These features are then used to predict the overall survival of the subjects. The main novelty in the proposed methods is the use of normalized brain parcellation data and tractography data from the human connectome project for analyzing MR images for segmentation and survival prediction. Experimental results are reported on the BraTS2018 data.