 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePLA-SGCN: Protein-Ligand Binding Affinity Prediction by Integrating Similar Pairs and Semi-supervised Graph Convolutional Network
May 13, 2024The protein-ligand binding affinity (PLA) prediction goal is to predict whether or not the ligand could bind to a protein sequence. Recently, in PLA prediction, deep learning has received much attention. Two steps are involved in deep learning-based approaches: feature extraction and task prediction step. Many deep learning-based approaches concentrate on introducing new feature extraction networks or integrating auxiliary knowledge like protein-protein interaction networks or gene ontology knowledge. Then, a task prediction network is designed simply using some fully connected layers. This paper aims to integrate retrieved similar hard protein-ligand pairs in PLA prediction (i.e., task prediction step) using a semi-supervised graph convolutional network (GCN). Hard protein-ligand pairs are retrieved for each input query sample based on the manifold smoothness constraint. Then, a graph is learned automatically in which each node is a protein-ligand pair, and each edge represents the similarity between pairs. In other words, an end-to-end framework is proposed that simultaneously retrieves hard similar samples, learns protein-ligand descriptor, learns the graph topology of the input sample with retrieved similar hard samples (learn adjacency matrix), and learns a semi-supervised GCN to predict the binding affinity (as task predictor). The training step adjusts the parameter values, and in the inference step, the learned model is fine-tuned for each input sample. To evaluate the proposed approach, it is applied to the four well-known PDBbind, Davis, KIBA, and BindingDB datasets. The results show that the proposed method significantly performs better than the comparable approaches.
HGTDR: Advancing Drug Repurposing with Heterogeneous Graph Transformers
May 12, 2024Motivation: Drug repurposing is a viable solution for reducing the time and cost associated with drug development. However, thus far, the proposed drug repurposing approaches still need to meet expectations. Therefore, it is crucial to offer a systematic approach for drug repurposing to achieve cost savings and enhance human lives. In recent years, using biological network-based methods for drug repurposing has generated promising results. Nevertheless, these methods have limitations. Primarily, the scope of these methods is generally limited concerning the size and variety of data they can effectively handle. Another issue arises from the treatment of heterogeneous data, which needs to be addressed or converted into homogeneous data, leading to a loss of information. A significant drawback is that most of these approaches lack end-to-end functionality, necessitating manual implementation and expert knowledge in certain stages. Results: We propose a new solution, HGTDR (Heterogeneous Graph Transformer for Drug Repurposing), to address the challenges associated with drug repurposing. HGTDR is a three-step approach for knowledge graph-based drug re-purposing: 1) constructing a heterogeneous knowledge graph, 2) utilizing a heterogeneous graph transformer network, and 3) computing relationship scores using a fully connected network. By leveraging HGTDR, users gain the ability to manipulate input graphs, extract information from diverse entities, and obtain their desired output. In the evaluation step, we demonstrate that HGTDR performs comparably to previous methods. Furthermore, we review medical studies to validate our method's top ten drug repurposing suggestions, which have exhibited promising results. We also demon-strated HGTDR's capability to predict other types of relations through numerical and experimental validation, such as drug-protein and disease-protein inter-relations.
CRISPR: Ensemble Model
Mar 05, 2024


Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) is a gene editing technology that has revolutionized the fields of biology and medicine. However, one of the challenges of using CRISPR is predicting the on-target efficacy and off-target sensitivity of single-guide RNAs (sgRNAs). This is because most existing methods are trained on separate datasets with different genes and cells, which limits their generalizability. In this paper, we propose a novel ensemble learning method for sgRNA design that is accurate and generalizable. Our method combines the predictions of multiple machine learning models to produce a single, more robust prediction. This approach allows us to learn from a wider range of data, which improves the generalizability of our model. We evaluated our method on a benchmark dataset of sgRNA designs and found that it outperformed existing methods in terms of both accuracy and generalizability. Our results suggest that our method can be used to design sgRNAs with high sensitivity and specificity, even for new genes or cells. This could have important implications for the clinical use of CRISPR, as it would allow researchers to design more effective and safer treatments for a variety of diseases.
Epsilon non-Greedy: A Bandit Approach for Unbiased Recommendation via Uniform Data
Oct 07, 2023



Often, recommendation systems employ continuous training, leading to a self-feedback loop bias in which the system becomes biased toward its previous recommendations. Recent studies have attempted to mitigate this bias by collecting small amounts of unbiased data. While these studies have successfully developed less biased models, they ignore the crucial fact that the recommendations generated by the model serve as the training data for subsequent training sessions. To address this issue, we propose a framework that learns an unbiased estimator using a small amount of uniformly collected data and focuses on generating improved training data for subsequent training iterations. To accomplish this, we view recommendation as a contextual multi-arm bandit problem and emphasize on exploring items that the model has a limited understanding of. We introduce a new offline sequential training schema that simulates real-world continuous training scenarios in recommendation systems, offering a more appropriate framework for studying self-feedback bias. We demonstrate the superiority of our model over state-of-the-art debiasing methods by conducting extensive experiments using the proposed training schema.
DANI: Fast Diffusion Aware Network Inference with Preserving Topological Structure Property
Oct 02, 2023The fast growth of social networks and their data access limitations in recent years has led to increasing difficulty in obtaining the complete topology of these networks. However, diffusion information over these networks is available, and many algorithms have been proposed to infer the underlying networks using this information. The previously proposed algorithms only focus on inferring more links and ignore preserving the critical topological characteristics of the underlying social networks. In this paper, we propose a novel method called DANI to infer the underlying network while preserving its structural properties. It is based on the Markov transition matrix derived from time series cascades, as well as the node-node similarity that can be observed in the cascade behavior from a structural point of view. In addition, the presented method has linear time complexity (increases linearly with the number of nodes, number of cascades, and square of the average length of cascades), and its distributed version in the MapReduce framework is also scalable. We applied the proposed approach to both real and synthetic networks. The experimental results showed that DANI has higher accuracy and lower run time while maintaining structural properties, including modular structure, degree distribution, connected components, density, and clustering coefficients, than well-known network inference methods.
ClusterSeq: Enhancing Sequential Recommender Systems with Clustering based Meta-Learning
Jul 25, 2023In practical scenarios, the effectiveness of sequential recommendation systems is hindered by the user cold-start problem, which arises due to limited interactions for accurately determining user preferences. Previous studies have attempted to address this issue by combining meta-learning with user and item-side information. However, these approaches face inherent challenges in modeling user preference dynamics, particularly for "minor users" who exhibit distinct preferences compared to more common or "major users." To overcome these limitations, we present a novel approach called ClusterSeq, a Meta-Learning Clustering-Based Sequential Recommender System. ClusterSeq leverages dynamic information in the user sequence to enhance item prediction accuracy, even in the absence of side information. This model preserves the preferences of minor users without being overshadowed by major users, and it capitalizes on the collective knowledge of users within the same cluster. Extensive experiments conducted on various benchmark datasets validate the effectiveness of ClusterSeq. Empirical results consistently demonstrate that ClusterSeq outperforms several state-of-the-art meta-learning recommenders. Notably, compared to existing meta-learning methods, our proposed approach achieves a substantial improvement of 16-39% in Mean Reciprocal Rank (MRR).
Domain Adaptation and Generalization on Functional Medical Images: A Systematic Survey
Dec 04, 2022



Machine learning algorithms have revolutionized different fields, including natural language processing, computer vision, signal processing, and medical data processing. Despite the excellent capabilities of machine learning algorithms in various tasks and areas, the performance of these models mainly deteriorates when there is a shift in the test and training data distributions. This gap occurs due to the violation of the fundamental assumption that the training and test data are independent and identically distributed (i.i.d). In real-world scenarios where collecting data from all possible domains for training is costly and even impossible, the i.i.d assumption can hardly be satisfied. The problem is even more severe in the case of medical images and signals because it requires either expensive equipment or a meticulous experimentation setup to collect data, even for a single domain. Additionally, the decrease in performance may have severe consequences in the analysis of medical records. As a result of such problems, the ability to generalize and adapt under distribution shifts (domain generalization (DG) and domain adaptation (DA)) is essential for the analysis of medical data. This paper provides the first systematic review of DG and DA on functional brain signals to fill the gap of the absence of a comprehensive study in this era. We provide detailed explanations and categorizations of datasets, approaches, and architectures used in DG and DA on functional brain images. We further address the attention-worthy future tracks in this field.
SCGG: A Deep Structure-Conditioned Graph Generative Model
Sep 20, 2022
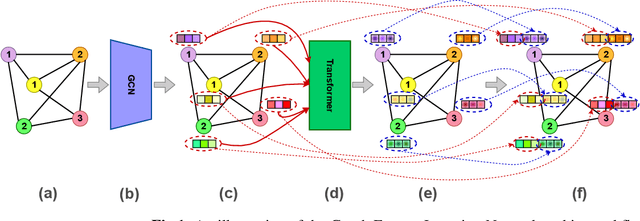
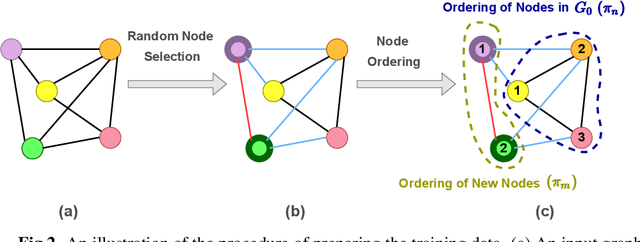
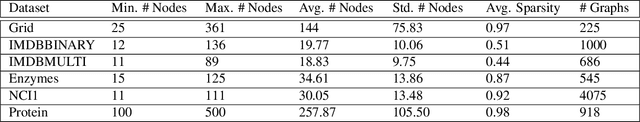
Deep learning-based graph generation approaches have remarkable capacities for graph data modeling, allowing them to solve a wide range of real-world problems. Making these methods able to consider different conditions during the generation procedure even increases their effectiveness by empowering them to generate new graph samples that meet the desired criteria. This paper presents a conditional deep graph generation method called SCGG that considers a particular type of structural conditions. Specifically, our proposed SCGG model takes an initial subgraph and autoregressively generates new nodes and their corresponding edges on top of the given conditioning substructure. The architecture of SCGG consists of a graph representation learning network and an autoregressive generative model, which is trained end-to-end. Using this model, we can address graph completion, a rampant and inherently difficult problem of recovering missing nodes and their associated edges of partially observed graphs. Experimental results on both synthetic and real-world datasets demonstrate the superiority of our method compared with state-of-the-art baselines.
SOInter: A Novel Deep Energy Based Interpretation Method for Explaining Structured Output Models
Feb 20, 2022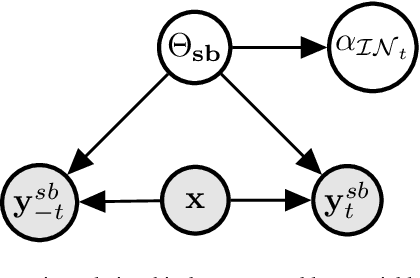
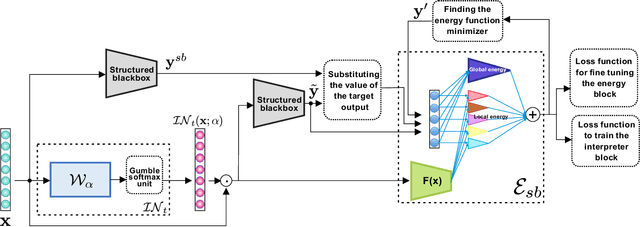
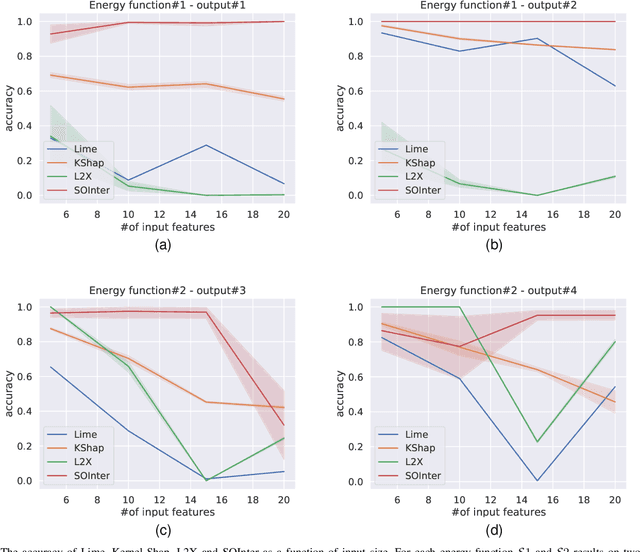
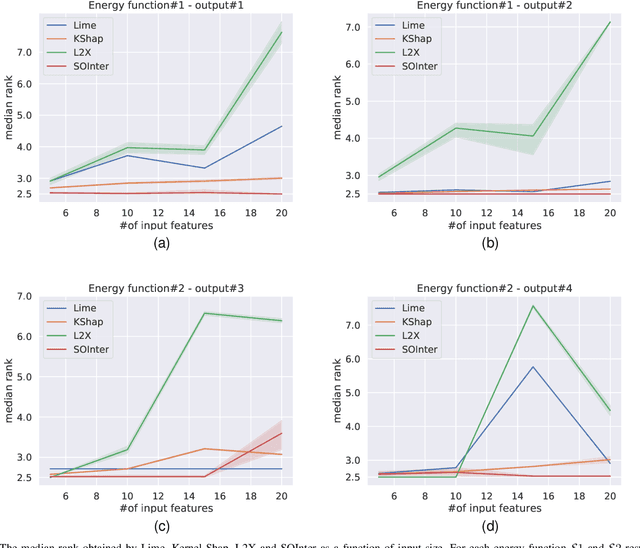
We propose a novel interpretation technique to explain the behavior of structured output models, which learn mappings between an input vector to a set of output variables simultaneously. Because of the complex relationship between the computational path of output variables in structured models, a feature can affect the value of output through other ones. We focus on one of the outputs as the target and try to find the most important features utilized by the structured model to decide on the target in each locality of the input space. In this paper, we assume an arbitrary structured output model is available as a black box and argue how considering the correlations between output variables can improve the explanation performance. The goal is to train a function as an interpreter for the target output variable over the input space. We introduce an energy-based training process for the interpreter function, which effectively considers the structural information incorporated into the model to be explained. The effectiveness of the proposed method is confirmed using a variety of simulated and real data sets.
LAP: An Attention-Based Module for Faithful Interpretation and Knowledge Injection in Convolutional Neural Networks
Jan 27, 2022
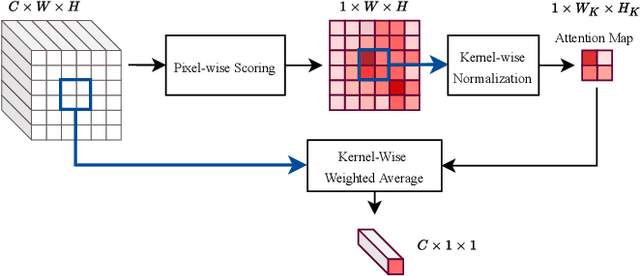
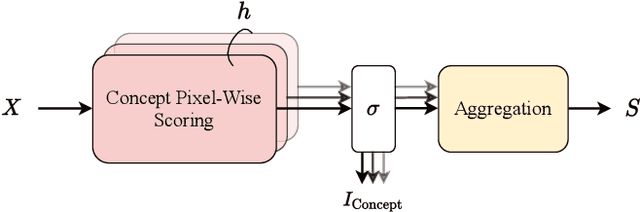

Despite the state-of-the-art performance of deep convolutional neural networks, they are susceptible to bias and malfunction in unseen situations. The complex computation behind their reasoning is not sufficiently human-understandable to develop trust. External explainer methods have tried to interpret the network decisions in a human-understandable way, but they are accused of fallacies due to their assumptions and simplifications. On the other side, the inherent self-interpretability of models, while being more robust to the mentioned fallacies, cannot be applied to the already trained models. In this work, we propose a new attention-based pooling layer, called Local Attention Pooling (LAP), that accomplishes self-interpretability and the possibility for knowledge injection while improving the model's performance. Moreover, several weakly-supervised knowledge injection methodologies are provided to enhance the process of training. We verified our claims by evaluating several LAP-extended models on three different datasets, including Imagenet. The proposed framework offers more valid human-understandable and more faithful-to-the-model interpretations than the commonly used white-box explainer methods.