 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerating scientific discovery with the common task framework
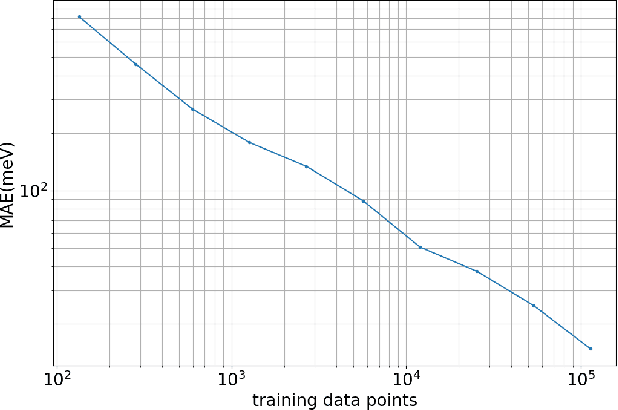
Nov 06, 2025Machine learning (ML) and artificial intelligence (AI) algorithms are transforming and empowering the characterization and control of dynamic systems in the engineering, physical, and biological sciences. These emerging modeling paradigms require comparative metrics to evaluate a diverse set of scientific objectives, including forecasting, state reconstruction, generalization, and control, while also considering limited data scenarios and noisy measurements. We introduce a common task framework (CTF) for science and engineering, which features a growing collection of challenge data sets with a diverse set of practical and common objectives. The CTF is a critically enabling technology that has contributed to the rapid advance of ML/AI algorithms in traditional applications such as speech recognition, language processing, and computer vision. There is a critical need for the objective metrics of a CTF to compare the diverse algorithms being rapidly developed and deployed in practice today across science and engineering.
TorchMD: A deep learning framework for molecular simulations
Dec 22, 2020


Molecular dynamics simulations provide a mechanistic description of molecules by relying on empirical potentials. The quality and transferability of such potentials can be improved leveraging data-driven models derived with machine learning approaches. Here, we present TorchMD, a framework for molecular simulations with mixed classical and machine learning potentials. All of force computations including bond, angle, dihedral, Lennard-Jones and Coulomb interactions are expressed as PyTorch arrays and operations. Moreover, TorchMD enables learning and simulating neural network potentials. We validate it using standard Amber all-atom simulations, learning an ab-initio potential, performing an end-to-end training and finally learning and simulating a coarse-grained model for protein folding. We believe that TorchMD provides a useful tool-set to support molecular simulations of machine learning potentials. Code and data are freely available at \url{github.com/torchmd}.
Deep Generative Markov State Models
May 19, 2018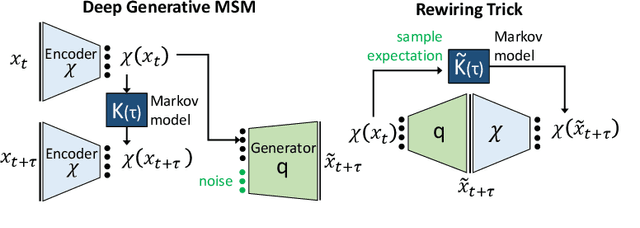

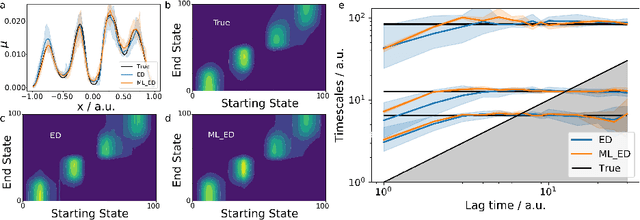
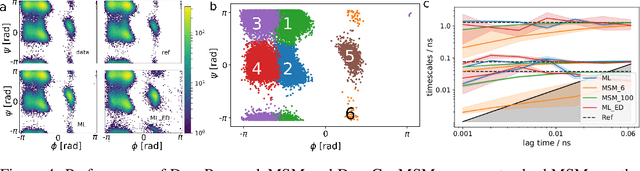
We propose a deep generative Markov State Model (DeepGenMSM) learning framework for inference of metastable dynamical systems and prediction of trajectories. After unsupervised training on time series data, the model contains (i) a probabilistic encoder that maps from high-dimensional configuration space to a small-sized vector indicating the membership to metastable (long-lived) states, (ii) a Markov chain that governs the transitions between metastable states and facilitates analysis of the long-time dynamics, and (iii) a generative part that samples the conditional distribution of configurations in the next time step. The model can be operated in a recursive fashion to generate trajectories to predict the system evolution from a defined starting state and propose new configurations. The DeepGenMSM is demonstrated to provide accurate estimates of the long-time kinetics and generate valid distributions for molecular dynamics (MD) benchmark systems. Remarkably, we show that DeepGenMSMs are able to make long time-steps in molecular configuration space and generate physically realistic structures in regions that were not seen in training data.