 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFlowSDR: Sufficient Dimension Reduction via Conditional Normalizing Flows
May 31, 2026Sufficient dimension reduction (SDR) seeks a low-dimensional linear projection of predictors that preserves the conditional distribution of the response. Existing methods target this conditional distribution indirectly, via inverse moments, local forward regression, or neural ensemble regression. We propose FlowSDR, a likelihood-based framework that jointly learns the projection and the conditional density by maximizing a conditional log-likelihood, with the density parameterized by monotone rational-quadratic spline flows. The estimator is Fisher consistent under the SDR model, and its sample objective admits a population interpretation in terms of mutual information. As a complementary model within the same likelihood framework, we introduce the neural Gaussian SDR, a heteroscedastic conditional Gaussian model whose mean and variance are parameterized by shared neural-network functions of the projected predictors. In simulations spanning Gaussian errors, heavy-tailed distributions, two-component mixtures, and settings with tail behavior not captured by mean-variance structure, FlowSDR recovers the central subspace more accurately than existing SDR methods and the neural Gaussian SDR baseline. We further validate these advantages on a face-age prediction task using the UTKFace dataset.
Wrapped Distributions on homogeneous Riemannian manifolds
Apr 20, 2022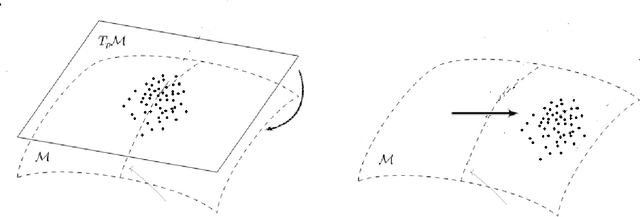

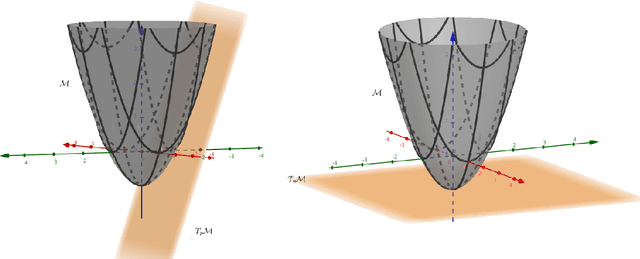

We provide a general framework for constructing probability distributions on Riemannian manifolds, taking advantage of area-preserving maps and isometries. Control over distributions' properties, such as parameters, symmetry and modality yield a family of flexible distributions that are straightforward to sample from, suitable for use within Monte Carlo algorithms and latent variable models, such as autoencoders. As an illustration, we empirically validate our approach by utilizing our proposed distributions within a variational autoencoder and a latent space network model. Finally, we take advantage of the generalized description of this framework to posit questions for future work.
Quantifying homologous proteins and proteoforms
Aug 05, 2017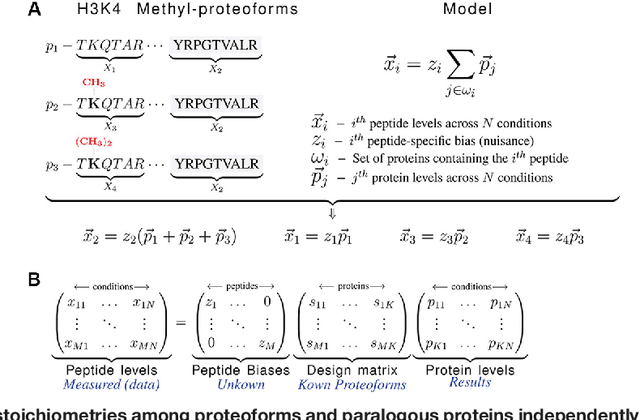
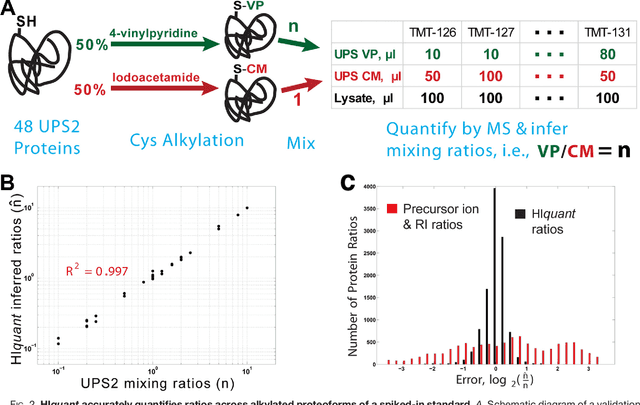
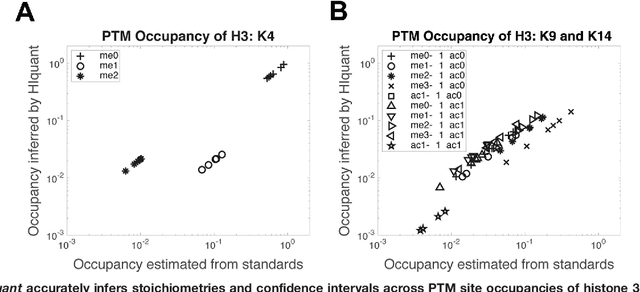
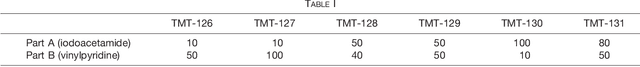
Many proteoforms - arising from alternative splicing, post-translational modifications (PTMs), or paralogous genes - have distinct biological functions, such as histone PTM proteoforms. However, their quantification by existing bottom-up mass-spectrometry (MS) methods is undermined by peptide-specific biases. To avoid these biases, we developed and implemented a first-principles model (HIquant) for quantifying proteoform stoichiometries. We characterized when MS data allow inferring proteoform stoichiometries by HIquant, derived an algorithm for optimal inference, and demonstrated experimentally high accuracy in quantifying fractional PTM occupancy without using external standards, even in the challenging case of the histone modification code. HIquant server is implemented at: https://web.northeastern.edu/slavov/2014_HIquant/
Inference of Network Summary Statistics Through Network Denoising
Dec 31, 2013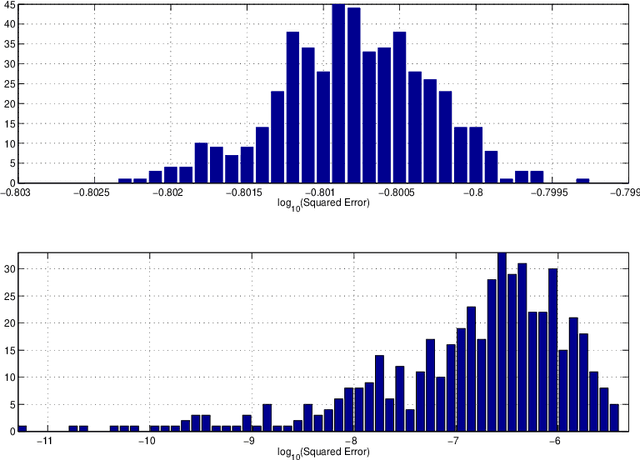

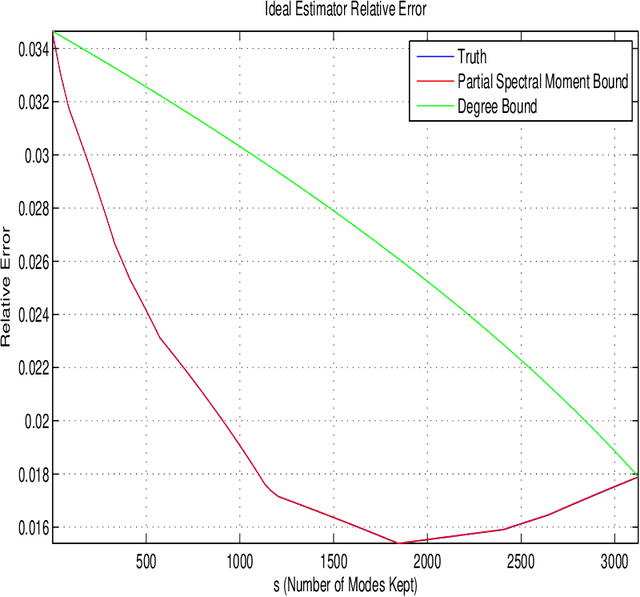
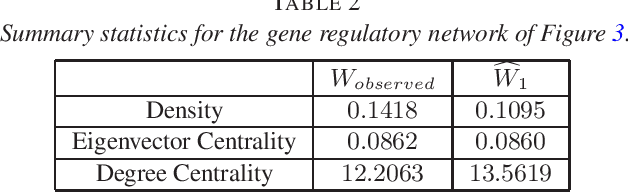
Consider observing an undirected network that is `noisy' in the sense that there are Type I and Type II errors in the observation of edges. Such errors can arise, for example, in the context of inferring gene regulatory networks in genomics or functional connectivity networks in neuroscience. Given a single observed network then, to what extent are summary statistics for that network representative of their analogues for the true underlying network? Can we infer such statistics more accurately by taking into account the noise in the observed network edges? In this paper, we answer both of these questions. In particular, we develop a spectral-based methodology using the adjacency matrix to `denoise' the observed network data and produce more accurate inference of the summary statistics of the true network. We characterize performance of our methodology through bounds on appropriate notions of risk in the $L^2$ sense, and conclude by illustrating the practical impact of this work on synthetic and real-world data.