 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph AI generates neurological hypotheses validated in molecular, organoid, and clinical systems
Dec 13, 2025Neurological diseases are the leading global cause of disability, yet most lack disease-modifying treatments. We present PROTON, a heterogeneous graph transformer that generates testable hypotheses across molecular, organoid, and clinical systems. To evaluate PROTON, we apply it to Parkinson's disease (PD), bipolar disorder (BD), and Alzheimer's disease (AD). In PD, PROTON linked genetic risk loci to genes essential for dopaminergic neuron survival and predicted pesticides toxic to patient-derived neurons, including the insecticide endosulfan, which ranked within the top 1.29% of predictions. In silico screens performed by PROTON reproduced six genome-wide $α$-synuclein experiments, including a split-ubiquitin yeast two-hybrid system (normalized enrichment score [NES] = 2.30, FDR-adjusted $p < 1 \times 10^{-4}$), an ascorbate peroxidase proximity labeling assay (NES = 2.16, FDR $< 1 \times 10^{-4}$), and a high-depth targeted exome sequencing study in 496 synucleinopathy patients (NES = 2.13, FDR $< 1 \times 10^{-4}$). In BD, PROTON predicted calcitriol as a candidate drug that reversed proteomic alterations observed in cortical organoids derived from BD patients. In AD, we evaluated PROTON predictions in health records from $n = 610,524$ patients at Mass General Brigham, confirming that five PROTON-predicted drugs were associated with reduced seven-year dementia risk (minimum hazard ratio = 0.63, 95% CI: 0.53-0.75, $p < 1 \times 10^{-7}$). PROTON generated neurological hypotheses that were evaluated across molecular, organoid, and clinical systems, defining a path for AI-driven discovery in neurological disease.
Quantifying homologous proteins and proteoforms
Aug 05, 2017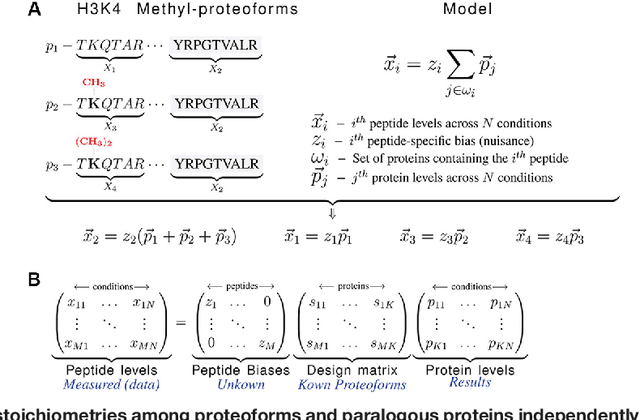
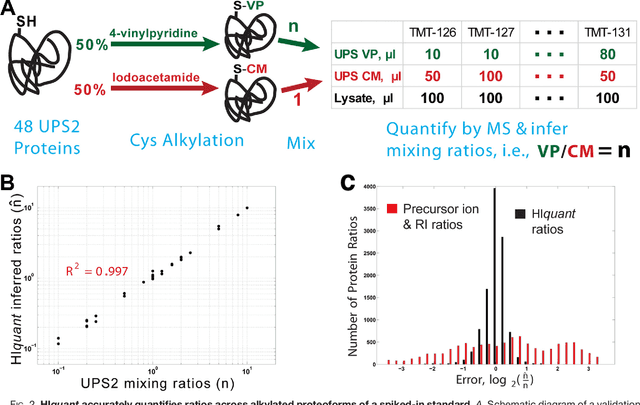
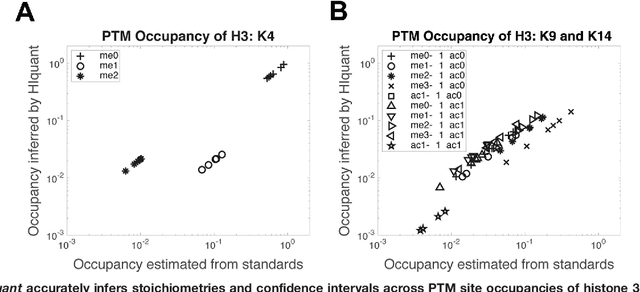
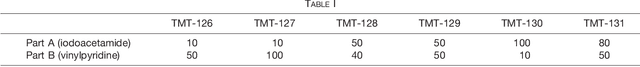
Many proteoforms - arising from alternative splicing, post-translational modifications (PTMs), or paralogous genes - have distinct biological functions, such as histone PTM proteoforms. However, their quantification by existing bottom-up mass-spectrometry (MS) methods is undermined by peptide-specific biases. To avoid these biases, we developed and implemented a first-principles model (HIquant) for quantifying proteoform stoichiometries. We characterized when MS data allow inferring proteoform stoichiometries by HIquant, derived an algorithm for optimal inference, and demonstrated experimentally high accuracy in quantifying fractional PTM occupancy without using external standards, even in the challenging case of the histone modification code. HIquant server is implemented at: https://web.northeastern.edu/slavov/2014_HIquant/