 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCase-Guided Sequential Assay Planning in Drug Discovery
Jan 21, 2026Optimally sequencing experimental assays in drug discovery is a high-stakes planning problem under severe uncertainty and resource constraints. A primary obstacle for standard reinforcement learning (RL) is the absence of an explicit environment simulator or transition data $(s, a, s')$; planning must rely solely on a static database of historical outcomes. We introduce the Implicit Bayesian Markov Decision Process (IBMDP), a model-based RL framework designed for such simulator-free settings. IBMDP constructs a case-guided implicit model of transition dynamics by forming a nonparametric belief distribution using similar historical outcomes. This mechanism enables Bayesian belief updating as evidence accumulates and employs ensemble MCTS planning to generate stable policies that balance information gain toward desired outcomes with resource efficiency. We validate IBMDP through comprehensive experiments. On a real-world central nervous system (CNS) drug discovery task, IBMDP reduced resource consumption by up to 92\% compared to established heuristics while maintaining decision confidence. To rigorously assess decision quality, we also benchmarked IBMDP in a synthetic environment with a computable optimal policy. Our framework achieves significantly higher alignment with this optimal policy than a deterministic value iteration alternative that uses the same similarity-based model, demonstrating the superiority of our ensemble planner. IBMDP offers a practical solution for sequential experimental design in data-rich but simulator-poor domains.
Transformer-Based Approach for Automated Functional Group Replacement in Chemical Compounds
Jan 12, 2026Functional group replacement is a pivotal approach in cheminformatics to enable the design of novel chemical compounds with tailored properties. Traditional methods for functional group removal and replacement often rely on rule-based heuristics, which can be limited in their ability to generate diverse and novel chemical structures. Recently, transformer-based models have shown promise in improving the accuracy and efficiency of molecular transformations, but existing approaches typically focus on single-step modeling, lacking the guarantee of structural similarity. In this work, we seek to advance the state of the art by developing a novel two-stage transformer model for functional group removal and replacement. Unlike one-shot approaches that generate entire molecules in a single pass, our method generates the functional group to be removed and appended sequentially, ensuring strict substructure-level modifications. Using a matched molecular pairs (MMPs) dataset derived from ChEMBL, we trained an encoder-decoder transformer model with SMIRKS-based representations to capture transformation rules effectively. Extensive evaluations demonstrate our method's ability to generate chemically valid transformations, explore diverse chemical spaces, and maintain scalability across varying search sizes.
Quantifying homologous proteins and proteoforms
Aug 05, 2017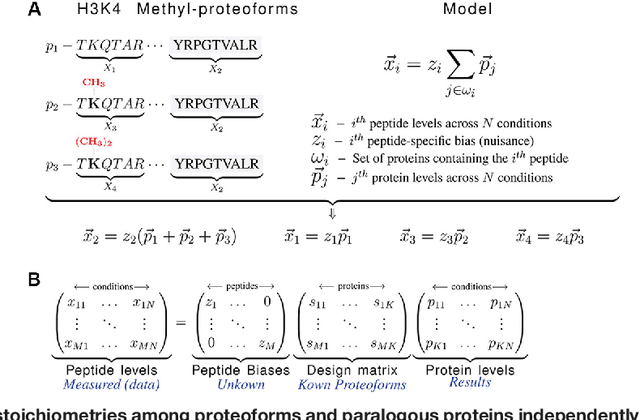
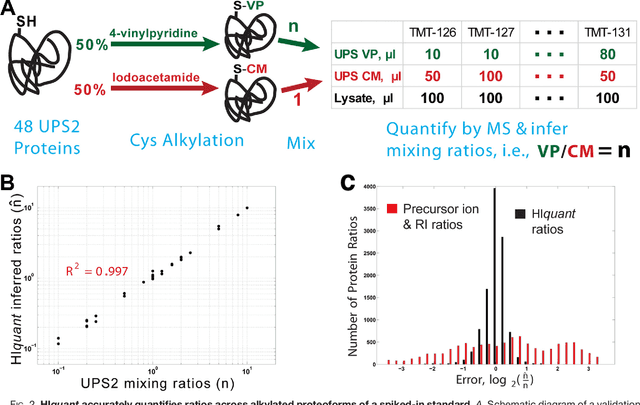
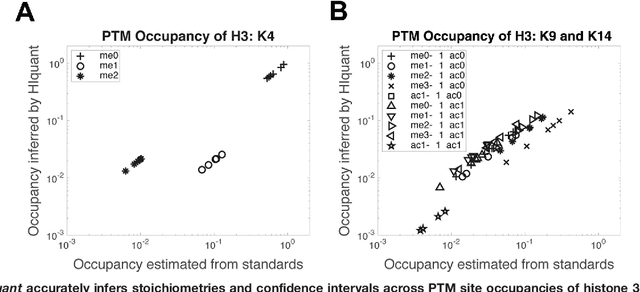
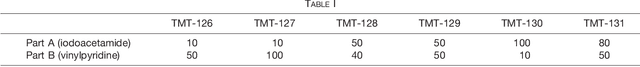
Many proteoforms - arising from alternative splicing, post-translational modifications (PTMs), or paralogous genes - have distinct biological functions, such as histone PTM proteoforms. However, their quantification by existing bottom-up mass-spectrometry (MS) methods is undermined by peptide-specific biases. To avoid these biases, we developed and implemented a first-principles model (HIquant) for quantifying proteoform stoichiometries. We characterized when MS data allow inferring proteoform stoichiometries by HIquant, derived an algorithm for optimal inference, and demonstrated experimentally high accuracy in quantifying fractional PTM occupancy without using external standards, even in the challenging case of the histone modification code. HIquant server is implemented at: https://web.northeastern.edu/slavov/2014_HIquant/