 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTranscriptomic Models for Immunotherapy Response Prediction Show Limited Cross-cohort Generalisability
Apr 07, 2026Immune checkpoint inhibitors (ICIs) have transformed cancer therapy; yet substantial proportion of patients exhibit intrinsic or acquired resistance, making accurate pre-treatment response prediction a critical unmet need. Transcriptomics-based biomarkers derived from bulk and single-cell RNA sequencing (scRNA-seq) offer a promising avenue for capturing tumour-immune interactions, yet the cross-cohort generalisability of existing prediction models remains unclear.We systematically benchmark nine state-of-the-art transcriptomic ICI response predictors, five bulk RNA-seq-based models (COMPASS, IRNet, NetBio, IKCScore, and TNBC-ICI) and four scRNA-seq-based models (PRECISE, DeepGeneX, Tres and scCURE), using publicly available independent datasets unseen during model development. Overall, predictive performance was modest: bulk RNA-seq models performed at or near chance level across most cohorts, while scRNA-seq models showed only marginal improvements. Pathway-level analyses revealed sparse and inconsistent biomarker signals across models. Although scRNA-seq-based predictors converged on immune-related programs such as allograft rejection, bulk RNA-seq-based models exhibited little reproducible overlap. PRECISE and NetBio identified the most coherent immune-related themes, whereas IRNet predominantly captured metabolic pathways weakly aligned with ICI biology. Together, these findings demonstrate the limited cross-cohort robustness and biological consistency of current transcriptomic ICI prediction models, underscoring the need for improved domain adaptation, standardised preprocessing, and biologically grounded model design.
Beyond Quantity: Trajectory Diversity Scaling for Code Agents
Feb 03, 2026As code large language models (LLMs) evolve into tool-interactive agents via the Model Context Protocol (MCP), their generalization is increasingly limited by low-quality synthetic data and the diminishing returns of quantity scaling. Moreover, quantity-centric scaling exhibits an early bottleneck that underutilizes trajectory data. We propose TDScaling, a Trajectory Diversity Scaling-based data synthesis framework for code agents that scales performance through diversity rather than raw volume. Under a fixed training budget, increasing trajectory diversity yields larger gains than adding more trajectories, improving the performance-cost trade-off for agent training. TDScaling integrates four innovations: (1) a Business Cluster mechanism that captures real-service logical dependencies; (2) a blueprint-driven multi-agent paradigm that enforces trajectory coherence; (3) an adaptive evolution mechanism that steers synthesis toward long-tail scenarios using Domain Entropy, Reasoning Mode Entropy, and Cumulative Action Complexity to prevent mode collapse; and (4) a sandboxed code tool that mitigates catastrophic forgetting of intrinsic coding capabilities. Experiments on general tool-use benchmarks (BFCL, tau^2-Bench) and code agent tasks (RebenchT, CodeCI, BIRD) demonstrate a win-win outcome: TDScaling improves both tool-use generalization and inherent coding proficiency. We plan to release the full codebase and the synthesized dataset (including 30,000+ tool clusters) upon publication.
RxSafeBench: Identifying Medication Safety Issues of Large Language Models in Simulated Consultation
Nov 06, 2025Numerous medical systems powered by Large Language Models (LLMs) have achieved remarkable progress in diverse healthcare tasks. However, research on their medication safety remains limited due to the lack of real world datasets, constrained by privacy and accessibility issues. Moreover, evaluation of LLMs in realistic clinical consultation settings, particularly regarding medication safety, is still underexplored. To address these gaps, we propose a framework that simulates and evaluates clinical consultations to systematically assess the medication safety capabilities of LLMs. Within this framework, we generate inquiry diagnosis dialogues with embedded medication risks and construct a dedicated medication safety database, RxRisk DB, containing 6,725 contraindications, 28,781 drug interactions, and 14,906 indication-drug pairs. A two-stage filtering strategy ensures clinical realism and professional quality, resulting in the benchmark RxSafeBench with 2,443 high-quality consultation scenarios. We evaluate leading open-source and proprietary LLMs using structured multiple choice questions that test their ability to recommend safe medications under simulated patient contexts. Results show that current LLMs struggle to integrate contraindication and interaction knowledge, especially when risks are implied rather than explicit. Our findings highlight key challenges in ensuring medication safety in LLM-based systems and provide insights into improving reliability through better prompting and task-specific tuning. RxSafeBench offers the first comprehensive benchmark for evaluating medication safety in LLMs, advancing safer and more trustworthy AI-driven clinical decision support.
SemanticST: Spatially Informed Semantic Graph Learning for Clustering, Integration, and Scalable Analysis of Spatial Transcriptomics
Jun 16, 2025Spatial transcriptomics (ST) technologies enable gene expression profiling with spatial resolution, offering unprecedented insights into tissue organization and disease heterogeneity. However, current analysis methods often struggle with noisy data, limited scalability, and inadequate modelling of complex cellular relationships. We present SemanticST, a biologically informed, graph-based deep learning framework that models diverse cellular contexts through multi-semantic graph construction. SemanticST builds multiple context-specific graphs capturing spatial proximity, gene expression similarity, and tissue domain structure, and learns disentangled embeddings for each. These are fused using an attention-inspired strategy to yield a unified, biologically meaningful representation. A community-aware min-cut loss improves robustness over contrastive learning, particularly in sparse ST data. SemanticST supports mini-batch training, making it the first graph neural network scalable to large-scale datasets such as Xenium (500,000 cells). Benchmarking across four platforms (Visium, Slide-seq, Stereo-seq, Xenium) and multiple human and mouse tissues shows consistent 20 percentage gains in ARI, NMI, and trajectory fidelity over DeepST, GraphST, and IRIS. In re-analysis of breast cancer Xenium data, SemanticST revealed rare and clinically significant niches, including triple receptor-positive clusters, spatially distinct DCIS-to-IDC transition zones, and FOXC2 tumour-associated myoepithelial cells, suggesting non-canonical EMT programs with stem-like features. SemanticST thus provides a scalable, interpretable, and biologically grounded framework for spatial transcriptomics analysis, enabling robust discovery across tissue types and diseases, and paving the way for spatially resolved tissue atlases and next-generation precision medicine.
Interpretable graph-based models on multimodal biomedical data integration: A technical review and benchmarking
May 03, 2025
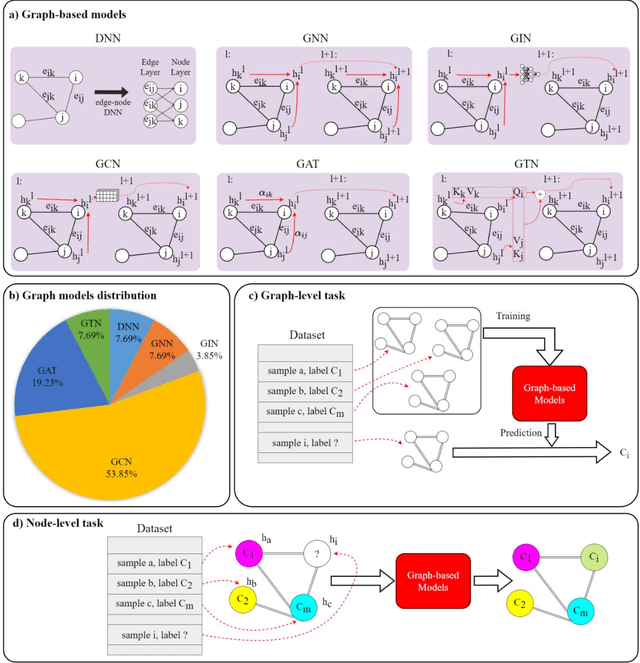
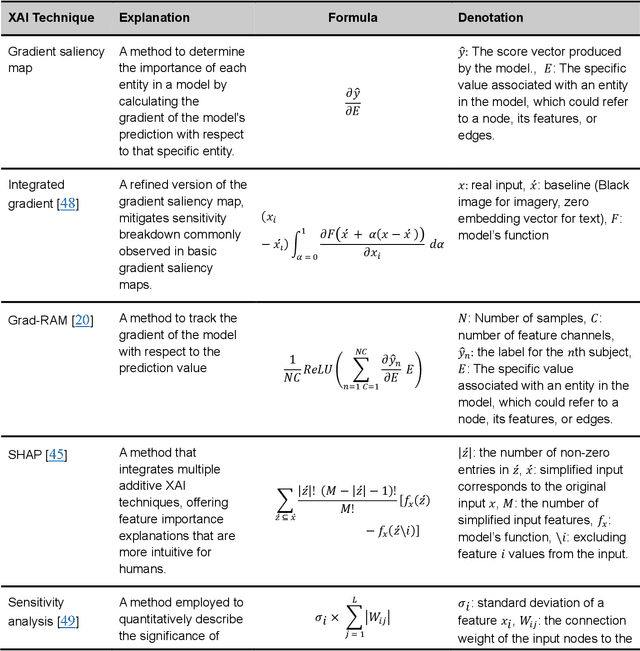
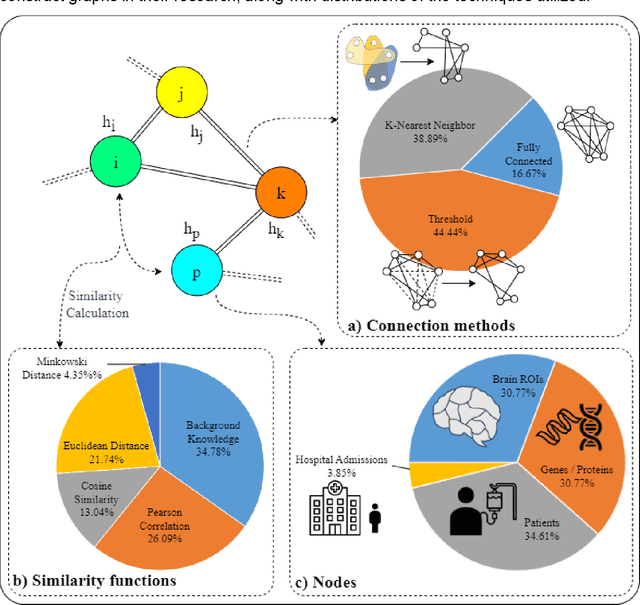
Integrating heterogeneous biomedical data including imaging, omics, and clinical records supports accurate diagnosis and personalised care. Graph-based models fuse such non-Euclidean data by capturing spatial and relational structure, yet clinical uptake requires regulator-ready interpretability. We present the first technical survey of interpretable graph based models for multimodal biomedical data, covering 26 studies published between Jan 2019 and Sep 2024. Most target disease classification, notably cancer and rely on static graphs from simple similarity measures, while graph-native explainers are rare; post-hoc methods adapted from non-graph domains such as gradient saliency, and SHAP predominate. We group existing approaches into four interpretability families, outline trends such as graph-in-graph hierarchies, knowledge-graph edges, and dynamic topology learning, and perform a practical benchmark. Using an Alzheimer disease cohort, we compare Sensitivity Analysis, Gradient Saliency, SHAP and Graph Masking. SHAP and Sensitivity Analysis recover the broadest set of known AD pathways and Gene-Ontology terms, whereas Gradient Saliency and Graph Masking surface complementary metabolic and transport signatures. Permutation tests show all four beat random gene sets, but with distinct trade-offs: SHAP and Graph Masking offer deeper biology at higher compute cost, while Gradient Saliency and Sensitivity Analysis are quicker though coarser. We also provide a step-by-step flowchart covering graph construction, explainer choice and resource budgeting to help researchers balance transparency and performance. This review synthesises the state of interpretable graph learning for multimodal medicine, benchmarks leading techniques, and charts future directions, from advanced XAI tools to under-studied diseases, serving as a concise reference for method developers and translational scientists.
xJailbreak: Representation Space Guided Reinforcement Learning for Interpretable LLM Jailbreaking
Jan 30, 2025



Safety alignment mechanism are essential for preventing large language models (LLMs) from generating harmful information or unethical content. However, cleverly crafted prompts can bypass these safety measures without accessing the model's internal parameters, a phenomenon known as black-box jailbreak. Existing heuristic black-box attack methods, such as genetic algorithms, suffer from limited effectiveness due to their inherent randomness, while recent reinforcement learning (RL) based methods often lack robust and informative reward signals. To address these challenges, we propose a novel black-box jailbreak method leveraging RL, which optimizes prompt generation by analyzing the embedding proximity between benign and malicious prompts. This approach ensures that the rewritten prompts closely align with the intent of the original prompts while enhancing the attack's effectiveness. Furthermore, we introduce a comprehensive jailbreak evaluation framework incorporating keywords, intent matching, and answer validation to provide a more rigorous and holistic assessment of jailbreak success. Experimental results show the superiority of our approach, achieving state-of-the-art (SOTA) performance on several prominent open and closed-source LLMs, including Qwen2.5-7B-Instruct, Llama3.1-8B-Instruct, and GPT-4o-0806. Our method sets a new benchmark in jailbreak attack effectiveness, highlighting potential vulnerabilities in LLMs. The codebase for this work is available at https://github.com/Aegis1863/xJailbreak.
ETAGE: Enhanced Test Time Adaptation with Integrated Entropy and Gradient Norms for Robust Model Performance
Sep 14, 2024



Test time adaptation (TTA) equips deep learning models to handle unseen test data that deviates from the training distribution, even when source data is inaccessible. While traditional TTA methods often rely on entropy as a confidence metric, its effectiveness can be limited, particularly in biased scenarios. Extending existing approaches like the Pseudo Label Probability Difference (PLPD), we introduce ETAGE, a refined TTA method that integrates entropy minimization with gradient norms and PLPD, to enhance sample selection and adaptation. Our method prioritizes samples that are less likely to cause instability by combining high entropy with high gradient norms out of adaptation, thus avoiding the overfitting to noise often observed in previous methods. Extensive experiments on CIFAR-10-C and CIFAR-100-C datasets demonstrate that our approach outperforms existing TTA techniques, particularly in challenging and biased scenarios, leading to more robust and consistent model performance across diverse test scenarios. The codebase for ETAGE is available on https://github.com/afsharshamsi/ETAGE.
Revolutionizing Genomics with Reinforcement Learning Techniques
Feb 26, 2023



In recent years, Reinforcement Learning (RL) has emerged as a powerful tool for solving a wide range of problems, including decision-making and genomics. The exponential growth of raw genomic data over the past two decades has exceeded the capacity of manual analysis, leading to a growing interest in automatic data analysis and processing. RL algorithms are capable of learning from experience with minimal human supervision, making them well-suited for genomic data analysis and interpretation. One of the key benefits of using RL is the reduced cost associated with collecting labeled training data, which is required for supervised learning. While there have been numerous studies examining the applications of Machine Learning (ML) in genomics, this survey focuses exclusively on the use of RL in various genomics research fields, including gene regulatory networks (GRNs), genome assembly, and sequence alignment. We present a comprehensive technical overview of existing studies on the application of RL in genomics, highlighting the strengths and limitations of these approaches. We then discuss potential research directions that are worthy of future exploration, including the development of more sophisticated reward functions as RL heavily depends on the accuracy of the reward function, the integration of RL with other machine learning techniques, and the application of RL to new and emerging areas in genomics research. Finally, we present our findings and conclude by summarizing the current state of the field and the future outlook for RL in genomics.