 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePre-training Molecular Graph Representation with 3D Geometry
Paper and Code
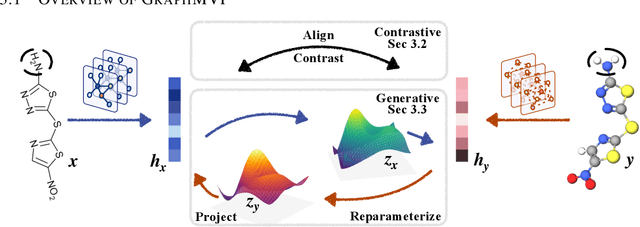
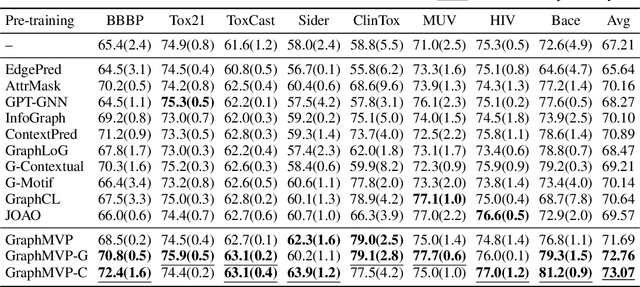

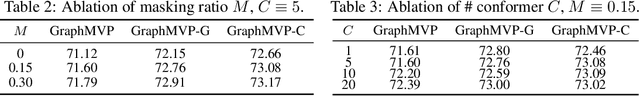
Molecular graph representation learning is a fundamental problem in modern drug and material discovery. Molecular graphs are typically modeled by their 2D topological structures, but it has been recently discovered that 3D geometric information plays a more vital role in predicting molecular functionalities. However, the lack of 3D information in real-world scenarios has significantly impeded the learning of geometric graph representation. To cope with this challenge, we propose the Graph Multi-View Pre-training (GraphMVP) framework where self-supervised learning (SSL) is performed by leveraging the correspondence and consistency between 2D topological structures and 3D geometric views. GraphMVP effectively learns a 2D molecular graph encoder that is enhanced by richer and more discriminative 3D geometry. We further provide theoretical insights to justify the effectiveness of GraphMVP. Finally, comprehensive experiments show that GraphMVP can consistently outperform existing graph SSL methods.