 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSCAN: Sparse Circuit Anchor Interpretable Neuron for Lifelong Knowledge Editing
Mar 16, 2026Large Language Models (LLMs) often suffer from catastrophic forgetting and collapse during sequential knowledge editing. This vulnerability stems from the prevailing dense editing paradigm, which treats models as black boxes and relies on coarse-grained parameter interventions that inevitably disrupt preserved knowledge. To address this, we propose SCAN (a sparse editing framework based on Sparse Circuit Anchored Neuron) which transforms editing into a mechanism-aware manipulation by constructing a knowledge circuit via Sparse Transcoders. Experiments on Gemma2, Qwen3, and Llama3.1 across CounterFact, ZsRE and WikiFactDiff demonstrate that SCAN achieves a superior performance, maintaining model integrity on benchmarks like MMLU and GSM8K even after 3,000 sequential edits, whereas other existing methods deteriorate progressively as editing accumulates, eventually resulting in model collapse.
REACT: Representation Extraction And Controllable Tuning to Overcome Overfitting in LLM Knowledge Editing
May 25, 2025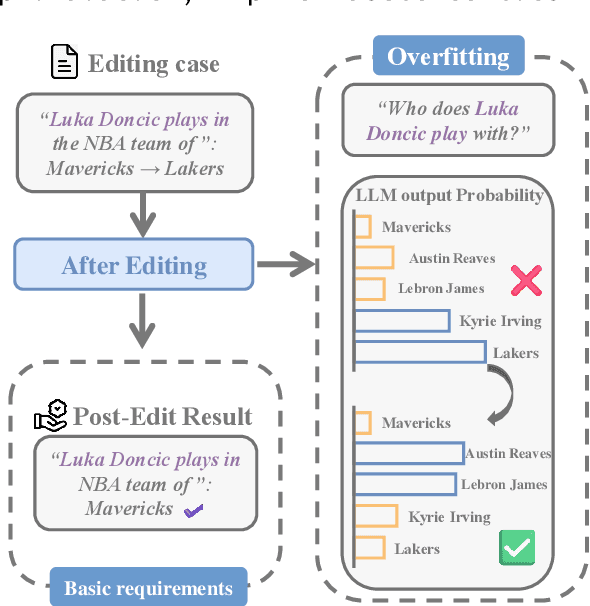
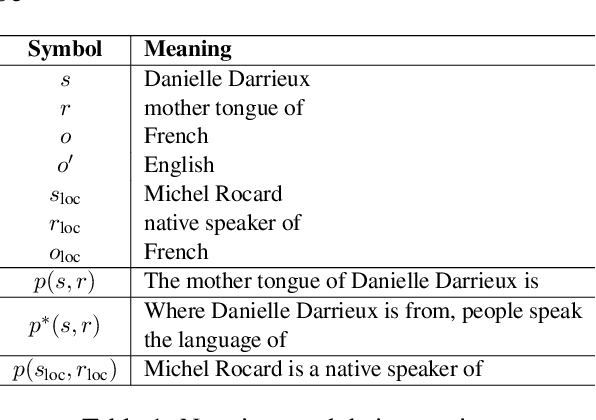
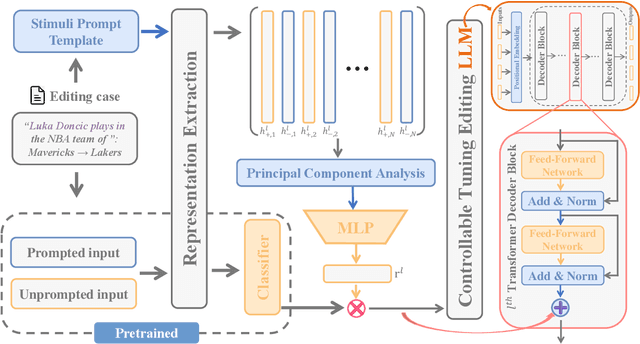
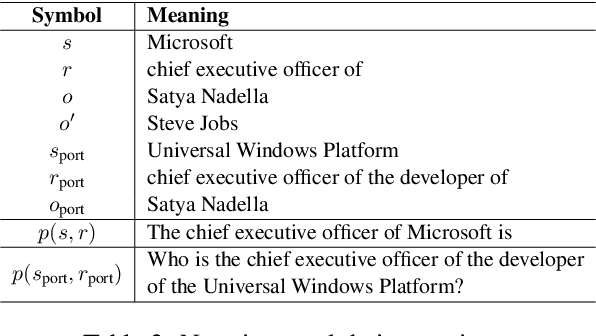
Large language model editing methods frequently suffer from overfitting, wherein factual updates can propagate beyond their intended scope, overemphasizing the edited target even when it's contextually inappropriate. To address this challenge, we introduce REACT (Representation Extraction And Controllable Tuning), a unified two-phase framework designed for precise and controllable knowledge editing. In the initial phase, we utilize tailored stimuli to extract latent factual representations and apply Principal Component Analysis with a simple learnbale linear transformation to compute a directional "belief shift" vector for each instance. In the second phase, we apply controllable perturbations to hidden states using the obtained vector with a magnitude scalar, gated by a pre-trained classifier that permits edits only when contextually necessary. Relevant experiments on EVOKE benchmarks demonstrate that REACT significantly reduces overfitting across nearly all evaluation metrics, and experiments on COUNTERFACT and MQuAKE shows that our method preserves balanced basic editing performance (reliability, locality, and generality) under diverse editing scenarios.
PDeepPP:A Deep learning framework with Pretrained Protein language for peptide classification
Feb 21, 2025Protein post-translational modifications (PTMs) and bioactive peptides (BPs) play critical roles in various biological processes and have significant therapeutic potential. However, identifying PTM sites and bioactive peptides through experimental methods is often labor-intensive, costly, and time-consuming. As a result, computational tools, particularly those based on deep learning, have become effective solutions for predicting PTM sites and peptide bioactivity. Despite progress in this field, existing methods still struggle with the complexity of protein sequences and the challenge of requiring high-quality predictions across diverse datasets. To address these issues, we propose a deep learning framework that integrates pretrained protein language models with a neural network combining transformer and CNN for peptide classification. By leveraging the ability of pretrained models to capture complex relationships within protein sequences, combined with the predictive power of parallel networks, our approach improves feature extraction while enhancing prediction accuracy. This framework was applied to multiple tasks involving PTM site and bioactive peptide prediction, utilizing large-scale datasets to enhance the model's robustness. In the comparison across 33 tasks, the model achieved state-of-the-art (SOTA) performance in 25 of them, surpassing existing methods and demonstrating its versatility across different datasets. Our results suggest that this approach provides a scalable and effective solution for large-scale peptide discovery and PTM analysis, paving the way for more efficient peptide classification and functional annotation.