 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSLOctolyzer: Fully automatic analysis toolkit for segmentation and feature extracting in scanning laser ophthalmoscopy images
Jun 24, 2024



Purpose: To describe SLOctolyzer: an open-source analysis toolkit for en face retinal vessels appearing in infrared reflectance scanning laser ophthalmoscopy (SLO) images. Methods: SLOctolyzer includes two main modules: segmentation and measurement. The segmentation module use deep learning methods to delineate retinal anatomy, while the measurement module quantifies key retinal vascular features such as vessel complexity, density, tortuosity, and calibre. We evaluate the segmentation module using unseen data and measure its reproducibility. Results: SLOctolyzer's segmentation module performed well against unseen internal test data (Dice for all-vessels, 0.9097; arteries, 0.8376; veins, 0.8525; optic disc, 0.9430; fovea, 0.8837). External validation against severe retinal pathology showed decreased performance (Dice for arteries, 0.7180; veins, 0.7470; optic disc, 0.9032). SLOctolyzer had good reproducibility (mean difference for fractal dimension, -0.0007; vessel density, -0.0003; vessel calibre, -0.3154 $\mu$m; tortuosity density, 0.0013). SLOctolyzer can process a macula-centred SLO image in under 20 seconds and a disc-centred SLO image in under 30 seconds using a standard laptop CPU. Conclusions: To our knowledge, SLOctolyzer is the first open-source tool to convert raw SLO images into reproducible and clinically meaningful retinal vascular parameters. SLO images are captured simultaneous to optical coherence tomography (OCT), and we believe our software will be useful for extracting retinal vascular measurements from large OCT image sets and linking them to ocular or systemic diseases. It requires no specialist knowledge or proprietary software, and allows manual correction of segmentations and re-computing of vascular metrics. SLOctolyzer is freely available at https://github.com/jaburke166/SLOctolyzer.
Automated and Network Structure Preserving Segmentation of Optical Coherence Tomography Angiograms
Dec 20, 2019
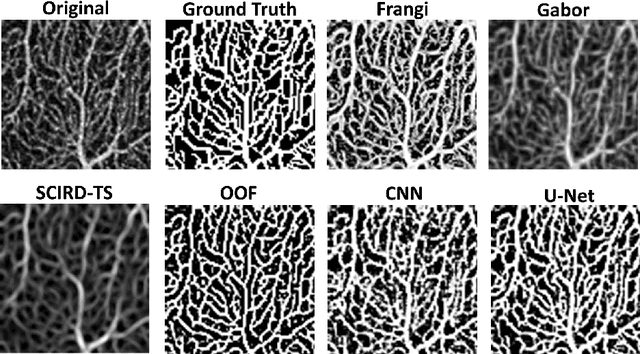

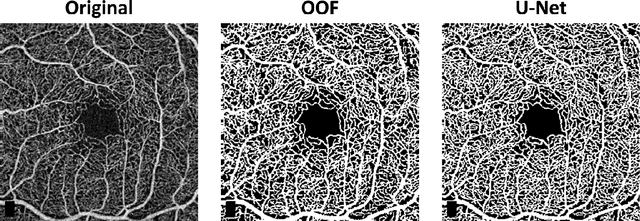
Optical coherence tomography angiography (OCTA) is a novel non-invasive imaging modality for the visualisation of microvasculature in vivo. OCTA has encountered broad adoption in retinal research. OCTA potential in the assessment of pathological conditions and the reproducibility of studies relies on the quality of the image analysis. However, automated segmentation of parafoveal OCTA images is still an open problem in the field. In this study, we generate the first open dataset of retinal parafoveal OCTA images with associated ground truth manual segmentations. Furthermore, we establish a standard for OCTA image segmentation by surveying a broad range of state-of-the-art vessel enhancement and binarisation procedures. We provide the most comprehensive comparison of these methods under a unified framework to date. Our results show that, for the set of images considered, the U-Net machine learning (ML) architecture achieves the best performance with a Dice similarity coefficient of 0.89. For applications where manually segmented data is not available to retrain this ML approach, our findings suggest that optimal oriented flux is the best handcrafted filter enhancement method for OCTA images from those considered. Furthermore, we report on the importance of preserving network connectivity in the segmentation to enable vascular network phenotyping. We introduce a new metric for network connectivity evaluations in segmented angiograms and report an accuracy of up to 0.94 in preserving the morphological structure of the network in our segmentations. Finally, we release our data and source code to support standardisation efforts in OCTA image segmentation.
Convolutional neural networks for structured omics: OmicsCNN and the OmicsConv layer
Oct 16, 2017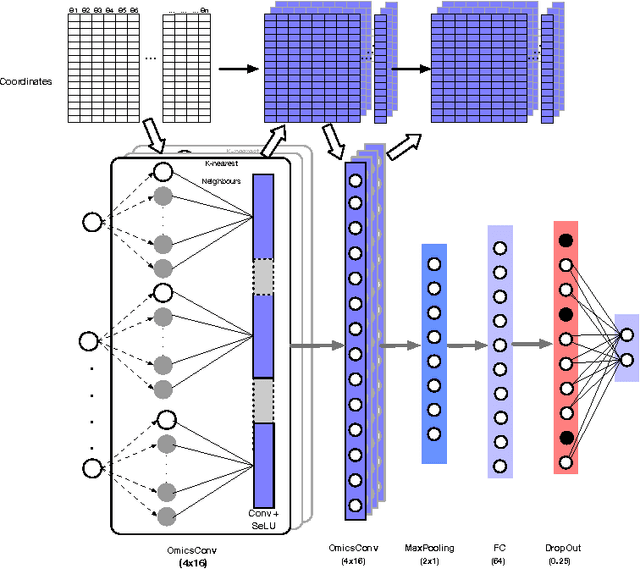
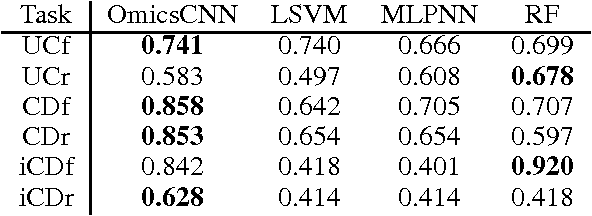
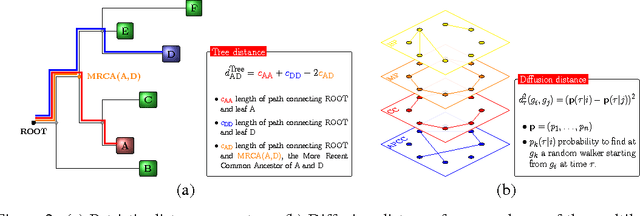
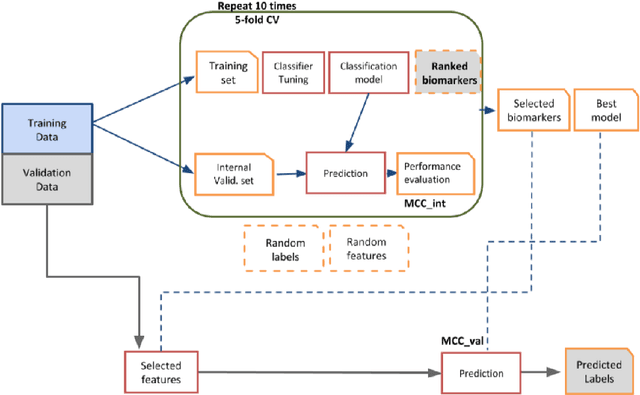
Convolutional Neural Networks (CNNs) are a popular deep learning architecture widely applied in different domains, in particular in classifying over images, for which the concept of convolution with a filter comes naturally. Unfortunately, the requirement of a distance (or, at least, of a neighbourhood function) in the input feature space has so far prevented its direct use on data types such as omics data. However, a number of omics data are metrizable, i.e., they can be endowed with a metric structure, enabling to adopt a convolutional based deep learning framework, e.g., for prediction. We propose a generalized solution for CNNs on omics data, implemented through a dedicated Keras layer. In particular, for metagenomics data, a metric can be derived from the patristic distance on the phylogenetic tree. For transcriptomics data, we combine Gene Ontology semantic similarity and gene co-expression to define a distance; the function is defined through a multilayer network where 3 layers are defined by the GO mutual semantic similarity while the fourth one by gene co-expression. As a general tool, feature distance on omics data is enabled by OmicsConv, a novel Keras layer, obtaining OmicsCNN, a dedicated deep learning framework. Here we demonstrate OmicsCNN on gut microbiota sequencing data, for Inflammatory Bowel Disease (IBD) 16S data, first on synthetic data and then a metagenomics collection of gut microbiota of 222 IBD patients.
Phylogenetic Convolutional Neural Networks in Metagenomics
Sep 06, 2017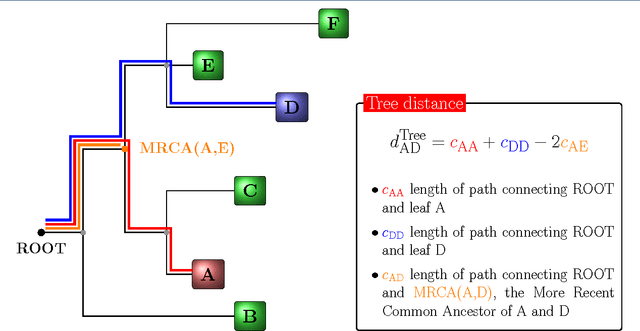
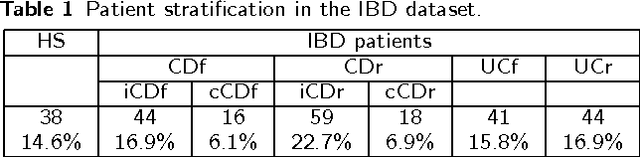
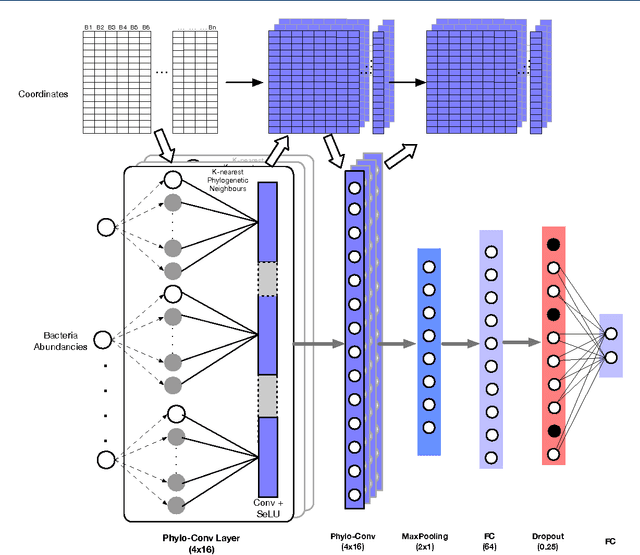
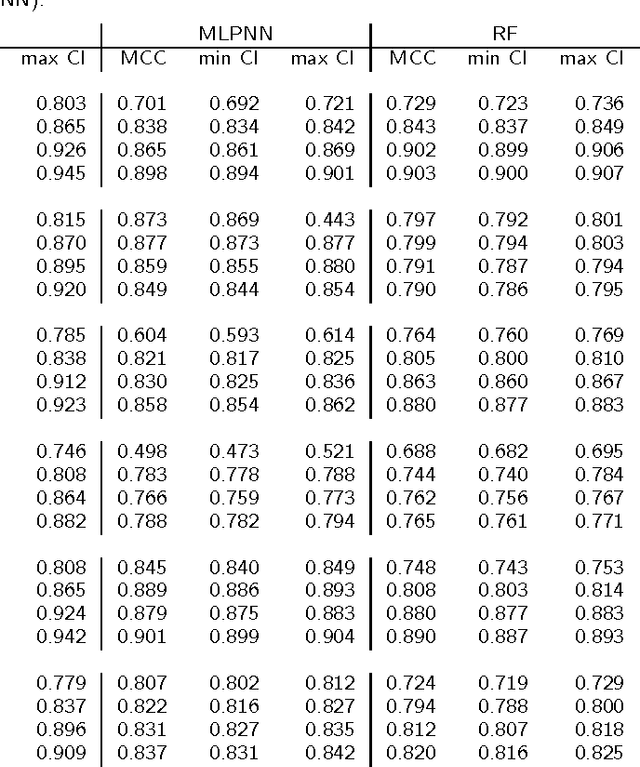
Background: Convolutional Neural Networks can be effectively used only when data are endowed with an intrinsic concept of neighbourhood in the input space, as is the case of pixels in images. We introduce here Ph-CNN, a novel deep learning architecture for the classification of metagenomics data based on the Convolutional Neural Networks, with the patristic distance defined on the phylogenetic tree being used as the proximity measure. The patristic distance between variables is used together with a sparsified version of MultiDimensional Scaling to embed the phylogenetic tree in a Euclidean space. Results: Ph-CNN is tested with a domain adaptation approach on synthetic data and on a metagenomics collection of gut microbiota of 38 healthy subjects and 222 Inflammatory Bowel Disease patients, divided in 6 subclasses. Classification performance is promising when compared to classical algorithms like Support Vector Machines and Random Forest and a baseline fully connected neural network, e.g. the Multi-Layer Perceptron. Conclusion: Ph-CNN represents a novel deep learning approach for the classification of metagenomics data. Operatively, the algorithm has been implemented as a custom Keras layer taking care of passing to the following convolutional layer not only the data but also the ranked list of neighbourhood of each sample, thus mimicking the case of image data, transparently to the user. Keywords: Metagenomics; Deep learning; Convolutional Neural Networks; Phylogenetic trees