 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStructure-aware Knowledge-guided Heterogeneous Mamba for Zygomaticomaxillary Suture Assessment
Jun 15, 2026The Zygomaticomaxillary Suture is a key circummaxillary structure that connects the zygomatic bone and the maxilla, which serves as a primary site of resistance during maxillary advancement, and its maturation status directly influences the timing and efficacy of orthopedic interventions. However, accurate staging of ZMS maturation remains challenging due to subtle high-frequency transitions in suture lines and the global semantic ambiguity between adjacent stages. To address this, we present the first public ZMS dataset, comprising 3,790 ZMS images covering the entire age range from 4 to 24 years. Based on this dataset, we propose SKMamba, a Structure-aware and Knowledge-guided Mamba-based multi-modal framework for automated ZMS maturation assessment. SKMamba adopts a decoupled dual-path architecture that mimics the hierarchical diagnostic process used by experienced orthodontists. We first introduce an Implicit Edge Extractor (IEE), which leverages structural pre-training to reduce trabecular noise and accentuate sutural boundaries. Complementarily, a Cross-Modal Semantic Alignment (CSA) module is designed to incorporate anatomical descriptions from a large language model (LLM). This module helps align local morphological cues with global semantic descriptions while ensuring that objective morphological evidence remains the primary basis for decisions. Extensive experiments on our ZMS dataset demonstrate that SKMamba achieves state-of-the-art performance compared to existing methods. Code is available at https://github.com/galaxygxq1116/SKMamba.
MC-NN: An End-to-End Multi-Channel Neural Network Approach for Predicting Influenza A Virus Hosts and Antigenic Types
Jun 08, 2023Influenza poses a significant threat to public health, particularly among the elderly, young children, and people with underlying dis-eases. The manifestation of severe conditions, such as pneumonia, highlights the importance of preventing the spread of influenza. An accurate and cost-effective prediction of the host and antigenic sub-types of influenza A viruses is essential to addressing this issue, particularly in resource-constrained regions. In this study, we propose a multi-channel neural network model to predict the host and antigenic subtypes of influenza A viruses from hemagglutinin and neuraminidase protein sequences. Our model was trained on a comprehensive data set of complete protein sequences and evaluated on various test data sets of complete and incomplete sequences. The results demonstrate the potential and practicality of using multi-channel neural networks in predicting the host and antigenic subtypes of influenza A viruses from both full and partial protein sequences.
Machine Learning for Flow Cytometry Data Analysis
Mar 16, 2023



Flow cytometry mainly used for detecting the characteristics of a number of biochemical substances based on the expression of specific markers in cells. It is particularly useful for detecting membrane surface receptors, antigens, ions, or during DNA/RNA expression. Not only can it be employed as a biomedical research tool for recognising distinctive types of cells in mixed populations, but it can also be used as a diagnostic tool for classifying abnormal cell populations connected with disease. Modern flow cytometers can rapidly analyse tens of thousands of cells at the same time while also measuring multiple parameters from a single cell. However, the rapid development of flow cytometers makes it challenging for conventional analysis methods to interpret flow cytometry data. Researchers need to be able to distinguish interesting-looking cell populations manually in multi-dimensional data collected from millions of cells. Thus, it is essential to find a robust approach for analysing flow cytometry data automatically, specifically in identifying cell populations automatically. This thesis mainly concerns discover the potential shortcoming of current automated-gating algorithms in both real datasets and synthetic datasets. Three representative automated clustering algorithms are selected to be applied, compared and evaluated by completely and partially automated gating. A subspace clustering ProClus also implemented in this thesis. The performance of ProClus in flow cytometry is not well, but it is still a useful algorithm to detect noise.
Dive into Machine Learning Algorithms for Influenza Virus Host Prediction with Hemagglutinin Sequences
Jul 28, 2022
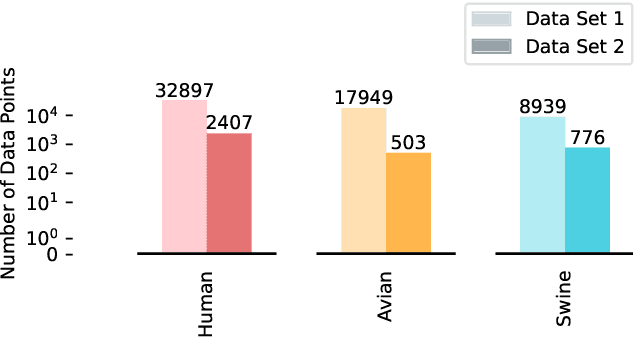
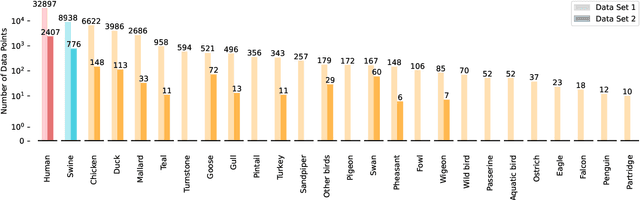

Influenza viruses mutate rapidly and can pose a threat to public health, especially to those in vulnerable groups. Throughout history, influenza A viruses have caused pandemics between different species. It is important to identify the origin of a virus in order to prevent the spread of an outbreak. Recently, there has been increasing interest in using machine learning algorithms to provide fast and accurate predictions for viral sequences. In this study, real testing data sets and a variety of evaluation metrics were used to evaluate machine learning algorithms at different taxonomic levels. As hemagglutinin is the major protein in the immune response, only hemagglutinin sequences were used and represented by position-specific scoring matrix and word embedding. The results suggest that the 5-grams-transformer neural network is the most effective algorithm for predicting viral sequence origins, with approximately 99.54% AUCPR, 98.01% F1 score and 96.60% MCC at a higher classification level, and approximately 94.74% AUCPR, 87.41% F1 score and 80.79% MCC at a lower classification level.
Multi-channel neural networks for predicting influenza A virus hosts and antigenic types
Jun 08, 2022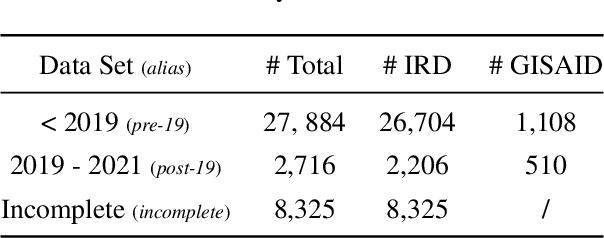
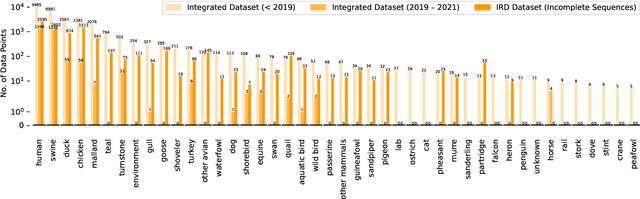

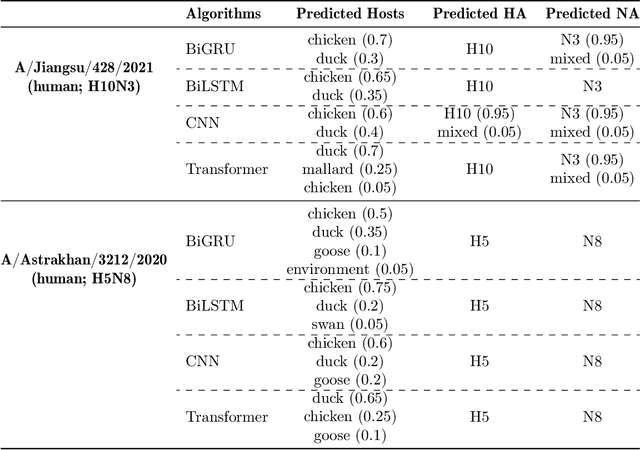
Influenza occurs every season and occasionally causes pandemics. Despite its low mortality rate, influenza is a major public health concern, as it can be complicated by severe diseases like pneumonia. A fast, accurate and low-cost method to predict the origin host and subtype of influenza viruses could help reduce virus transmission and benefit resource-poor areas. In this work, we propose multi-channel neural networks to predict antigenic types and hosts of influenza A viruses with hemagglutinin and neuraminidase protein sequences. An integrated data set containing complete protein sequences were used to produce a pre-trained model, and two other data sets were used for testing the model's performance. One test set contained complete protein sequences, and another test set contained incomplete protein sequences. The results suggest that multi-channel neural networks are applicable and promising for predicting influenza A virus hosts and antigenic subtypes with complete and partial protein sequences.
Predicting Influenza A Viral Host Using PSSM and Word Embeddings
Jan 04, 2022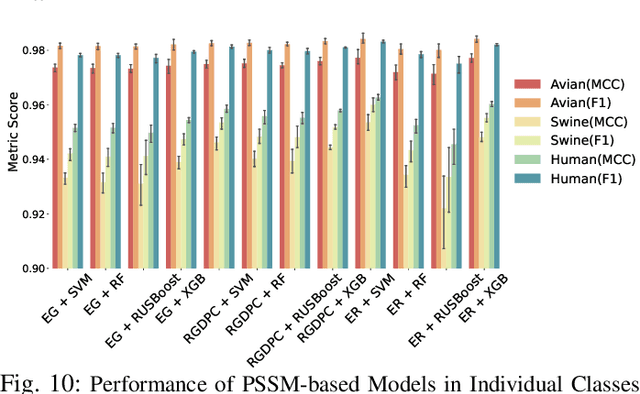
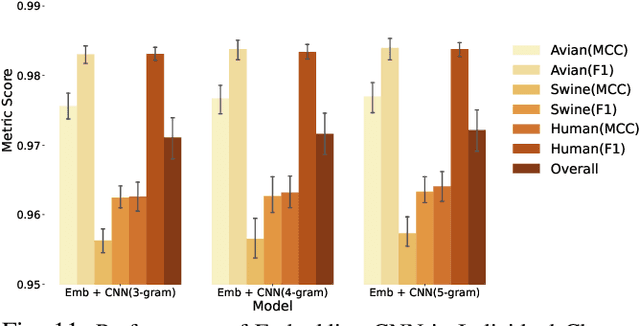
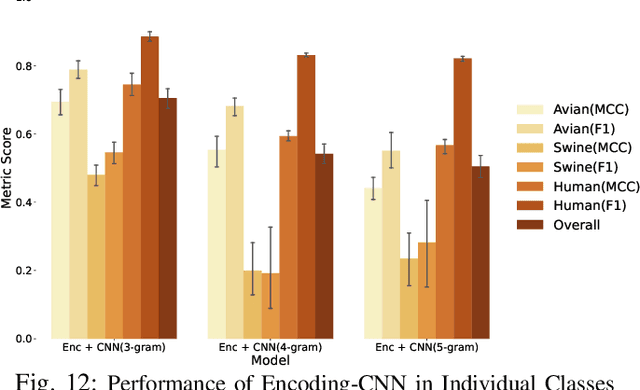
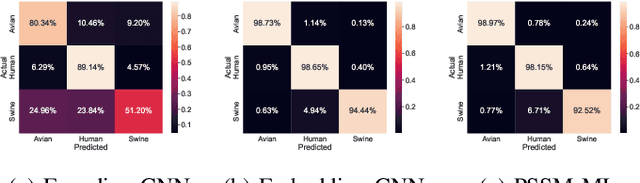
The rapid mutation of the influenza virus threatens public health. Reassortment among viruses with different hosts can lead to a fatal pandemic. However, it is difficult to detect the original host of the virus during or after an outbreak as influenza viruses can circulate between different species. Therefore, early and rapid detection of the viral host would help reduce the further spread of the virus. We use various machine learning models with features derived from the position-specific scoring matrix (PSSM) and features learned from word embedding and word encoding to infer the origin host of viruses. The results show that the performance of the PSSM-based model reaches the MCC around 95%, and the F1 around 96%. The MCC obtained using the model with word embedding is around 96%, and the F1 is around 97%.
* Accepted for publication at CIBCB 2021. V1: accepted version + minor correction to table 1