 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtDAT: A Unified Framework for Protein Sequence Design from Any Protein Text Description
Dec 05, 2024



Protein design has become a critical method in advancing significant potential for various applications such as drug development and enzyme engineering. However, protein design methods utilizing large language models with solely pretraining and fine-tuning struggle to capture relationships in multi-modal protein data. To address this, we propose ProtDAT, a de novo fine-grained framework capable of designing proteins from any descriptive protein text input. ProtDAT builds upon the inherent characteristics of protein data to unify sequences and text as a cohesive whole rather than separate entities. It leverages an innovative multi-modal cross-attention, integrating protein sequences and textual information for a foundational level and seamless integration. Experimental results demonstrate that ProtDAT achieves the state-of-the-art performance in protein sequence generation, excelling in rationality, functionality, structural similarity, and validity. On 20,000 text-sequence pairs from Swiss-Prot, it improves pLDDT by 6%, TM-score by 0.26, and reduces RMSD by 1.2 {\AA}, highlighting its potential to advance protein design.
FLIER: Few-shot Language Image Models Embedded with Latent Representations
Oct 10, 2024



As the boosting development of large vision-language models like Contrastive Language-Image Pre-training (CLIP), many CLIP-like methods have shown impressive abilities on visual recognition, especially in low-data regimes scenes. However, we have noticed that most of these methods are limited to introducing new modifications on text and image encoder. Recently, latent diffusion models (LDMs) have shown good ability on image generation. The potent capabilities of LDMs direct our focus towards the latent representations sampled by UNet. Inspired by the conjecture in CoOp that learned prompts encode meanings beyond the existing vocabulary, we assume that, for deep models, the latent representations are concise and accurate understanding of images, in which high-frequency, imperceptible details are abstracted away. In this paper, we propose a Few-shot Language Image model Embedded with latent Representations (FLIER) for image recognition by introducing a latent encoder jointly trained with CLIP's image encoder, it incorporates pre-trained vision-language knowledge of CLIP and the latent representations from Stable Diffusion. We first generate images and corresponding latent representations via Stable Diffusion with the textual inputs from GPT-3. With latent representations as "models-understandable pixels", we introduce a flexible convolutional neural network with two convolutional layers to be the latent encoder, which is simpler than most encoders in vision-language models. The latent encoder is jointly trained with CLIP's image encoder, transferring pre-trained knowledge to downstream tasks better. Experiments and extensive ablation studies on various visual classification tasks demonstrate that FLIER performs state-of-the-art on 11 datasets for most few-shot classification.
Molecule Generation for Drug Design: a Graph Learning Perspective
Feb 18, 2022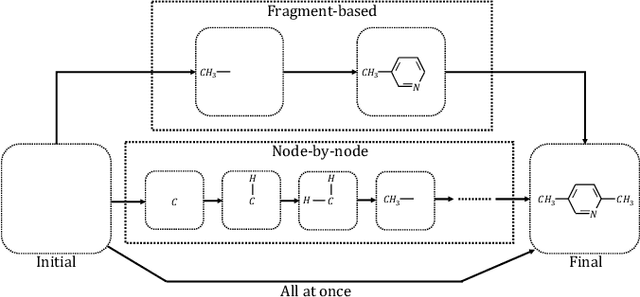
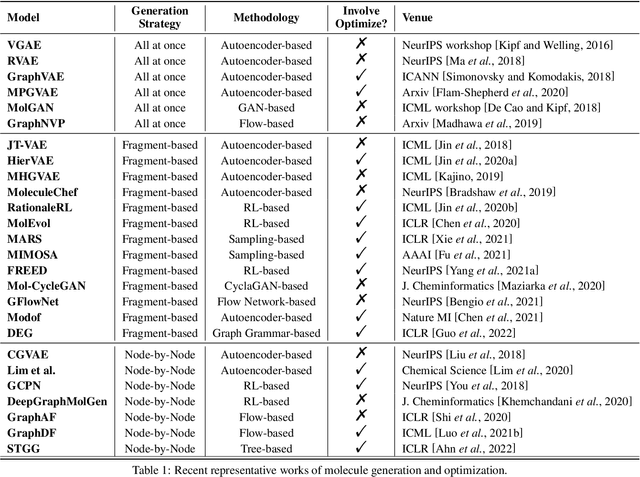
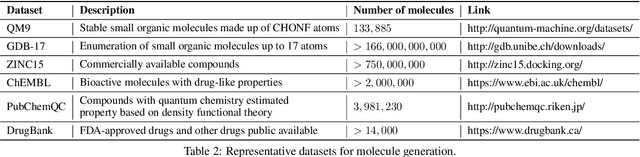
Machine learning has revolutionized many fields, and graph learning is recently receiving increasing attention. From the application perspective, one of the emerging and attractive areas is aiding the design and discovery of molecules, especially in drug industry. In this survey, we provide an overview of the state-of-the-art molecule (and mostly for de novo drug) design and discovery aiding methods whose methodology involves (deep) graph learning. Specifically, we propose to categorize these methods into three groups: i) all at once, ii) fragment-based and iii) node-by-node. We further present some representative public datasets and summarize commonly utilized evaluation metrics for generation and optimization, respectively. Finally, we discuss challenges and directions for future research, from the drug design perspective.
Attention based convolutional neural network for predicting RNA-protein binding sites
Dec 06, 2017

RNA-binding proteins (RBPs) play crucial roles in many biological processes, e.g. gene regulation. Computational identification of RBP binding sites on RNAs are urgently needed. In particular, RBPs bind to RNAs by recognizing sequence motifs. Thus, fast locating those motifs on RNA sequences is crucial and time-efficient for determining whether the RNAs interact with the RBPs or not. In this study, we present an attention based convolutional neural network, iDeepA, to predict RNA-protein binding sites from raw RNA sequences. We first encode RNA sequences into one-hot encoding. Next, we design a deep learning model with a convolutional neural network (CNN) and an attention mechanism, which automatically search for important positions, e.g. binding motifs, to learn discriminant high-level features for predicting RBP binding sites. We evaluate iDeepA on publicly gold-standard RBP binding sites derived from CLIP-seq data. The results demonstrate iDeepA achieves comparable performance with other state-of-the-art methods.