 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeModeling Atomic Conformational Ensembles of Proteins via Test-Time Supervision of Boltz-2 on Cryo-EM Density Maps
May 11, 2026Knowledge of a protein's atomic conformational ensemble is critical to determining its function, yet state-of-the-art ensemble prediction models are limited by lack of high-quality conformational data from simulation or experiment. Recent advances in heterogeneous reconstruction for cryo-electron microscopy (cryo-EM) have enabled scientists to visualize ensembles of density maps for larger proteins and complexes not typically accessible through simulation, but building atomic models into these maps remains a challenge. Traditionally, ensemble prediction models are trained via a two-stage process: experimental density maps are converted into atomic structural ensembles through model building, after which these structures are used to train sequence-to-atomic ensemble predictors. In this work, we propose a new principle for fine-tuning pre-trained static structure prediction models such as Boltz-2 directly on raw cryo-EM maps, bypassing the two-stage process. We apply this technique to the problem of atomic model building by fine-tuning Boltz-2 to generate atomic conformations from an input ensemble of cryo-EM maps, achieving superior model building accuracy compared to prior work. Beyond overfitting to individual map ensembles, our method, CryoSampler, also shows preliminary evidence of in-domain generalization after fine-tuning, sampling diverse atomic conformations for an unseen sequences within the same protein family without requiring cryo-EM data. These capabilities indicate that CryoSampler holds the potential to train next-generation atomic ensemble prediction models directly on raw cryo-EM measurements.
MIMIC: A Generative Multimodal Foundation Model for Biomolecules
Apr 27, 2026Biological function emerges from coupled constraints across sequence, structure, regulation, evolution, and cellular context, yet most foundation models in biology are trained within one modality or for a fixed forward task. We present MIMIC, a generative multimodal foundation model trained on our newly curated and aligned dataset, LORE, linking nucleic acid, protein, evolutionary, structural, regulatory, and semantic/contextual modalities within partially observed biomolecular states. MIMIC uses a split-track encoder-decoder architecture to condition on arbitrary subsets of observed modalities and reconstruct or generate missing components of molecular state across the genome, transcriptome, and proteome. Multimodal conditioning consistently improves MIMIC's sequence reconstruction relative to sequence-only inputs, while its learned representations enable state-of-the-art performance on RNA and protein downstream tasks. MIMIC achieves state-of-the-art splicing prediction, and its joint generative formulation enables isoform-aware inference that further improves performance. Beyond prediction, the same generative framework supports constrained design. For RNA, MIMIC identifies corrective edits in a clinically relevant HBB splice-disrupting mutation without reverting it by using evolutionary and structural signals. For proteins, jointly conditioning on shape and surface chemistry of PD-L1 and hACE2 binding sites produces diverse, high-confidence sequences with strong in silico support for target binding. Finally, MIMIC uses experimental context as semantic conditioning to model assay-dependent RNA chemical probing, rather than treating context as a fixed output. Together, these results position MIMIC's aligned multimodal generative modeling as a strong foundation for unifying representation learning, conditional prediction, and constrained biomolecular design within a single model.
CryoBench: Diverse and challenging datasets for the heterogeneity problem in cryo-EM
Aug 10, 2024



Cryo-electron microscopy (cryo-EM) is a powerful technique for determining high-resolution 3D biomolecular structures from imaging data. As this technique can capture dynamic biomolecular complexes, 3D reconstruction methods are increasingly being developed to resolve this intrinsic structural heterogeneity. However, the absence of standardized benchmarks with ground truth structures and validation metrics limits the advancement of the field. Here, we propose CryoBench, a suite of datasets, metrics, and performance benchmarks for heterogeneous reconstruction in cryo-EM. We propose five datasets representing different sources of heterogeneity and degrees of difficulty. These include conformational heterogeneity generated from simple motions and random configurations of antibody complexes and from tens of thousands of structures sampled from a molecular dynamics simulation. We also design datasets containing compositional heterogeneity from mixtures of ribosome assembly states and 100 common complexes present in cells. We then perform a comprehensive analysis of state-of-the-art heterogeneous reconstruction tools including neural and non-neural methods and their sensitivity to noise, and propose new metrics for quantitative comparison of methods. We hope that this benchmark will be a foundational resource for analyzing existing methods and new algorithmic development in both the cryo-EM and machine learning communities.
Exploring the Adjugate Matrix Approach to Quaternion Pose Extraction
May 17, 2022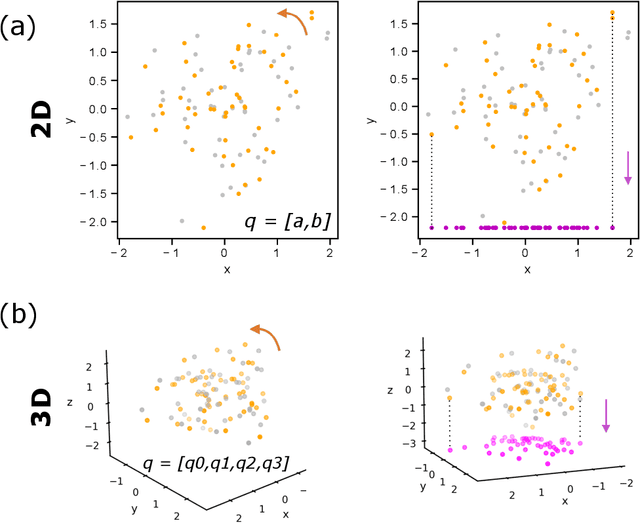
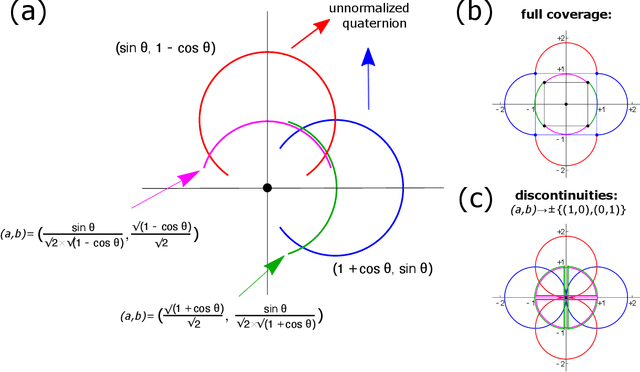
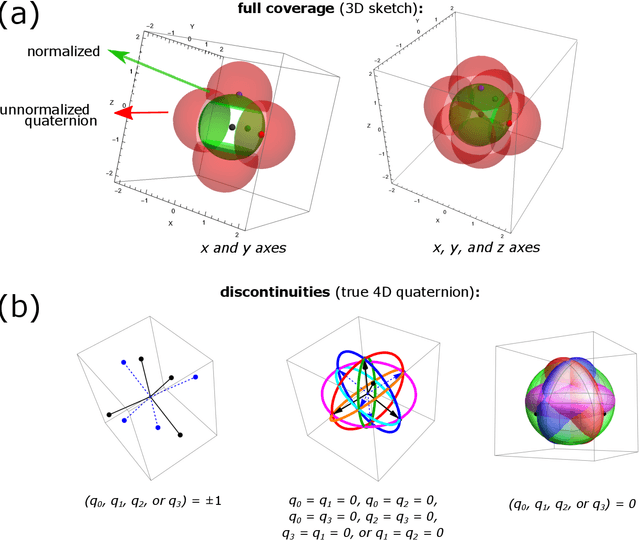
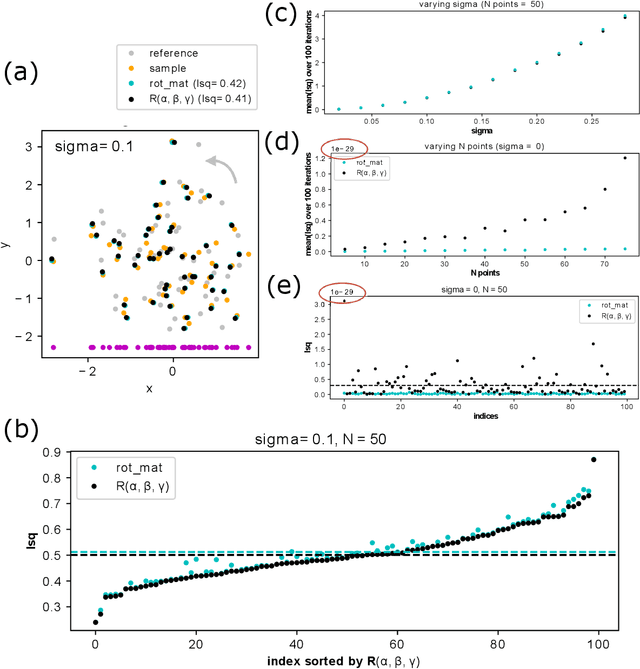
Quaternions are important for a wide variety of rotation-related problems in computer graphics, machine vision, and robotics. We study the nontrivial geometry of the relationship between quaternions and rotation matrices by exploiting the adjugate matrix of the characteristic equation of a related eigenvalue problem to obtain the manifold of the space of a quaternion eigenvector. We argue that quaternions parameterized by their corresponding rotation matrices cannot be expressed, for example, in machine learning tasks, as single-valued functions: the quaternion solution must instead be treated as a manifold, with different algebraic solutions for each of several single-valued sectors represented by the adjugate matrix. We conclude with novel constructions exploiting the quaternion adjugate variables to revisit several classic pose estimation applications: 2D point-cloud matching, 2D point-cloud-to-projection matching, 3D point-cloud matching, 3D orthographic point-cloud-to-projection matching, and 3D perspective point-cloud-to-projection matching. We find an exact solution to the 3D orthographic least squares pose extraction problem, and apply it successfully also to the perspective pose extraction problem with results that improve on existing methods.