 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMILD-Net: Minimal Information Loss Dilated Network for Gland Instance Segmentation in Colon Histology Images
Jun 07, 2018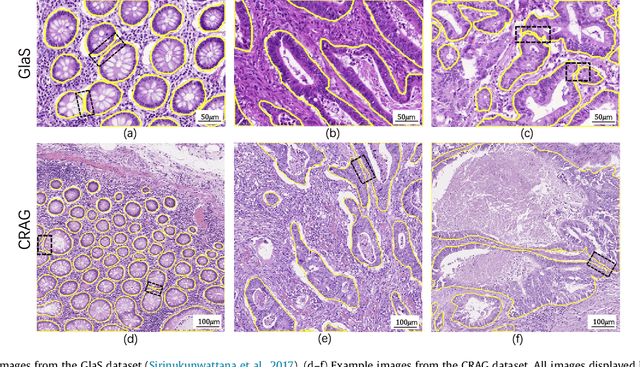
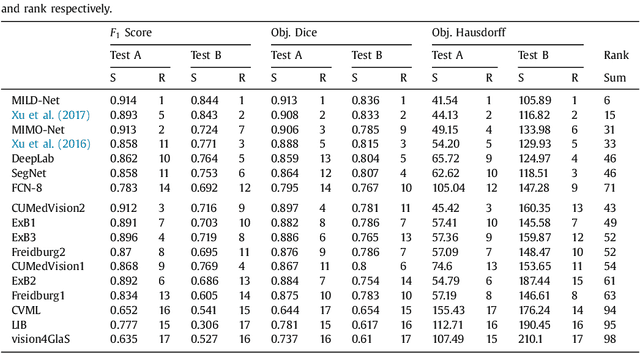
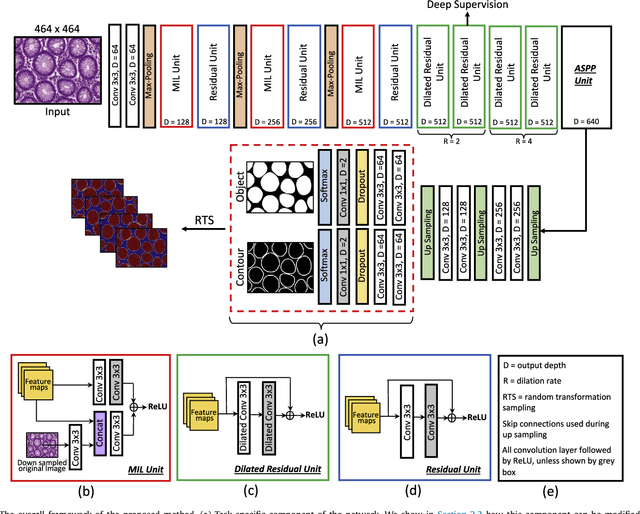
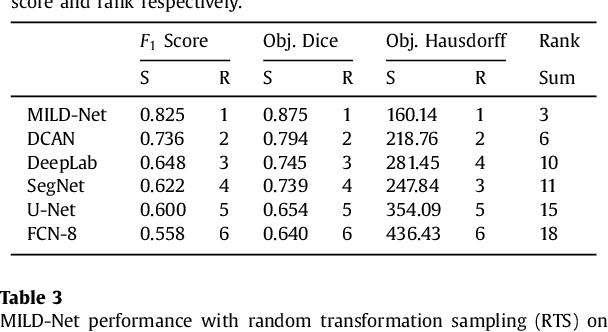
The analysis of glandular morphology within colon histopathology images is a crucial step in determining the stage of colon cancer. Despite the importance of this task, manual segmentation is laborious, time-consuming and can suffer from subjectivity among pathologists. The rise of computational pathology has led to the development of automated methods for gland segmentation that aim to overcome the challenges of manual segmentation. However, this task is non-trivial due to the large variability in glandular appearance and the difficulty in differentiating between certain glandular and non-glandular histological structures. Furthermore, within pathological practice, a measure of uncertainty is essential for diagnostic decision making. For example, ambiguous areas may require further examination from numerous pathologists. To address these challenges, we propose a fully convolutional neural network that counters the loss of information caused by max-pooling by re-introducing the original image at multiple points within the network. We also use atrous spatial pyramid pooling with varying dilation rates for resolution maintenance and multi-level aggregation. To incorporate uncertainty, we introduce random transformations during test time for an enhanced segmentation result that simultaneously generates an uncertainty map, highlighting areas of ambiguity. We show that this map can be used to define a metric for disregarding predictions with high uncertainty. The proposed network achieves state-of-the-art performance on the GlaS challenge dataset, as part of MICCAI 2015, and on a second independent colorectal adenocarcinoma dataset.
Micro-Net: A unified model for segmentation of various objects in microscopy images
Apr 22, 2018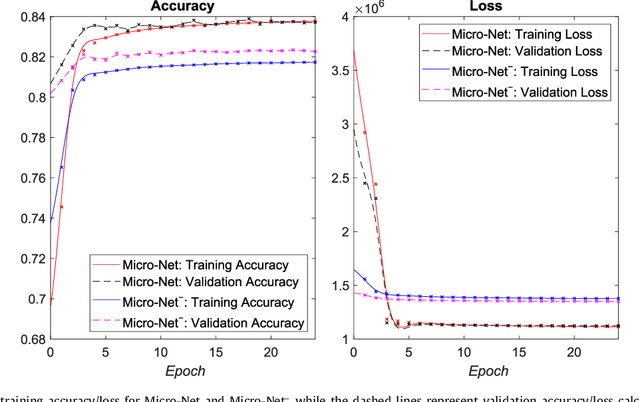
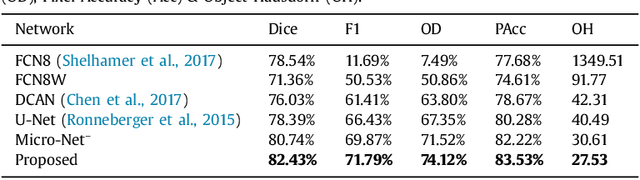
Object segmentation and structure localization are important steps in automated image analysis pipelines for microscopy images. We present a convolution neural network (CNN) based deep learning architecture for segmentation of objects in microscopy images. The proposed network can be used to segment cells, nuclei and glands in fluorescence microscopy and histology images after slight tuning of its parameters. It trains itself at multiple resolutions of the input image, connects the intermediate layers for better localization and context and generates the output using multi-resolution deconvolution filters. The extra convolutional layers which bypass the max-pooling operation allow the network to train for variable input intensities and object size and make it robust to noisy data. We compare our results on publicly available data sets and show that the proposed network outperforms the state-of-the-art.