 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeComputational framework for multistep metabolic pathway design
Apr 15, 2026In silico tools are important for generating novel hypotheses and exploring alternatives in de novo metabolic pathway design. However, while many computational frameworks have been proposed for retrobiosynthesis, few successful examples of algorithm-guided xenobiotic biochemical retrosynthesis have been reported in the literature. Deep learning has improved the quality of synthesis and retrosynthesis in organic chemistry applications. Inspired by this progress, we explored combining deep learning of biochemical transformations with the traditional retrobiosynthetic workflow to improve in silico synthetic metabolic pathway designs. To develop our computational biosynthetic pathway design framework, we assembled metabolic reaction and enzymatic template data from public databases. A data augmentation procedure, adapted from literature, was carried out to enrich the assembled reaction dataset with artificial metabolic reactions generated by enzymatic reaction templates. Two neural network-based pathway ranking models were trained as binary classifiers to distinguish assembled reactions from artificial counterparts; each model output a scalar quantifying the plausibility of a 1-step or 2-step pathway. Combining these two models with enzymatic templates, we built a multistep retrobiosynthesis pipeline and validated it by reproducing some natural and non-natural pathways computationally.
Retrieval-Augmented Foundation Models for Matched Molecular Pair Transformations to Recapitulate Medicinal Chemistry Intuition
Feb 18, 2026Matched molecular pairs (MMPs) capture the local chemical edits that medicinal chemists routinely use to design analogs, but existing ML approaches either operate at the whole-molecule level with limited edit controllability or learn MMP-style edits from restricted settings and small models. We propose a variable-to-variable formulation of analog generation and train a foundation model on large-scale MMP transformations (MMPTs) to generate diverse variables conditioned on an input variable. To enable practical control, we develop prompting mechanisms that let the users specify preferred transformation patterns during generation. We further introduce MMPT-RAG, a retrieval-augmented framework that uses external reference analogs as contextual guidance to steer generation and generalize from project-specific series. Experiments on general chemical corpora and patent-specific datasets demonstrate improved diversity, novelty, and controllability, and show that our method recovers realistic analog structures in practical discovery scenarios.
PepThink-R1: LLM for Interpretable Cyclic Peptide Optimization with CoT SFT and Reinforcement Learning
Aug 20, 2025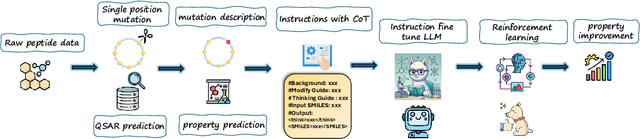
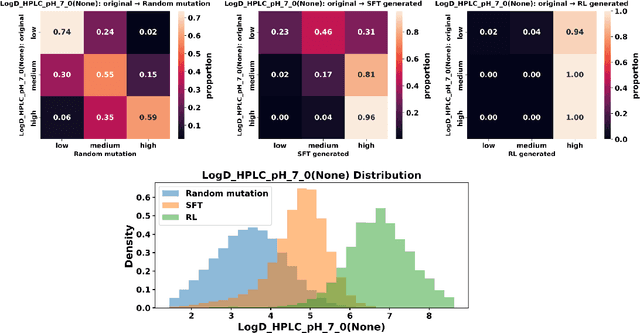
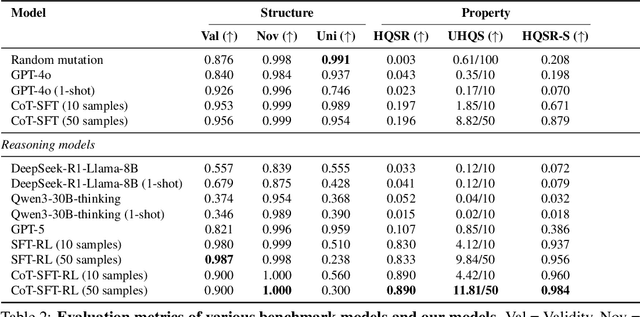
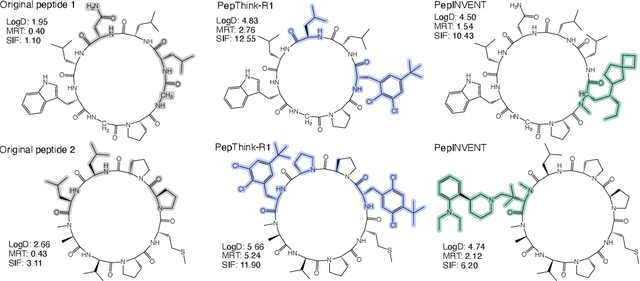
Designing therapeutic peptides with tailored properties is hindered by the vastness of sequence space, limited experimental data, and poor interpretability of current generative models. To address these challenges, we introduce PepThink-R1, a generative framework that integrates large language models (LLMs) with chain-of-thought (CoT) supervised fine-tuning and reinforcement learning (RL). Unlike prior approaches, PepThink-R1 explicitly reasons about monomer-level modifications during sequence generation, enabling interpretable design choices while optimizing for multiple pharmacological properties. Guided by a tailored reward function balancing chemical validity and property improvements, the model autonomously explores diverse sequence variants. We demonstrate that PepThink-R1 generates cyclic peptides with significantly enhanced lipophilicity, stability, and exposure, outperforming existing general LLMs (e.g., GPT-5) and domain-specific baseline in both optimization success and interpretability. To our knowledge, this is the first LLM-based peptide design framework that combines explicit reasoning with RL-driven property control, marking a step toward reliable and transparent peptide optimization for therapeutic discovery.