 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeData-Driven Soft Labeling Scales DNA Read Classification to Whole-Body Cell-Type Deconvolution
Jul 06, 2026Cell-type deconvolution, the task of estimating the proportions of constituent cell types in a heterogeneous biological sample, is a core problem in computational biology. Methods that rely on epigenetic marks such as DNA methylation typically operate on aggregated methylation estimates, discarding the pattern-level information carried by individual DNA reads. Existing read-level approaches that exploit this information are scarce, and all remain restricted to few-class settings; scaling them further is an open problem because, at scale, non-discriminative reads dominate and hard labels conflict with the many-to-many mapping between methylation patterns and cell types, preventing classifier convergence. To overcome this, we propose data-driven soft labels that estimate the conditional cell-type distribution for each read, and integrate this scheme into Syto, a new modular framework for read-level classification-based deconvolution. On a whole-body atlas of 39 human cell types, Syto reduces MSE by 2.56$\times$ over SoTA, with gains transferring to an out-of-distribution dataset spanning 16 tissues. Syto lays the foundation for modeling increasingly large cell-type panels, with improved applications in biology and healthcare. The proposed soft-labeling scheme is further translatable to any setting with a many-to-many signal-to-label mapping.
Querying Counterfactuals on Tissue Graphs with Supervised Disentanglement
Jun 07, 2026\textit{Tissue graph counterfactuals} ask how a cell's expression would change under altered spatial neighbor contexts. Such queries are central to predicting cell behavior in tissues, but lack a unified definition, with existing methods targeting specific intervention types or treating cells as i.i.d. In this work, we first formalize \textit{tissue graph counterfactuals} as a class of spatial interventions that either rewire connections between cells (\textit{edge perturbation}) or modify the expression of their neighbors (\textit{node perturbation}). We then introduce \textit{Cellina} {\renewcommand{\thefootnote}‡\footnote{https://cellina.readthedocs.io}\addtocounter{footnote}{-1}}, a framework that uses supervised disentanglement to decompose a cell's intrinsic state from its spatial context, using the latter as a conditioning input for counterfactual predictions. Across benchmarks spanning over 2.5 million spatially-resolved cells in colorectal cancer and mouse brain, \textit{Cellina} outperforms spatially-informed and non-spatial competitors in tissue perturbations, disentanglement, and scalability. Additionally, we show that \textit{Cellina} reveals biologically distinct cancer subdomains in an unsupervised manner and enables targeted neighbor perturbation simulations.
Matrix factorization with Binary Components
Jan 23, 2014
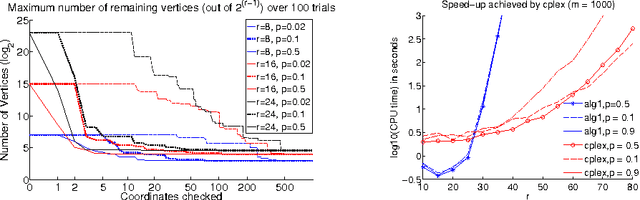
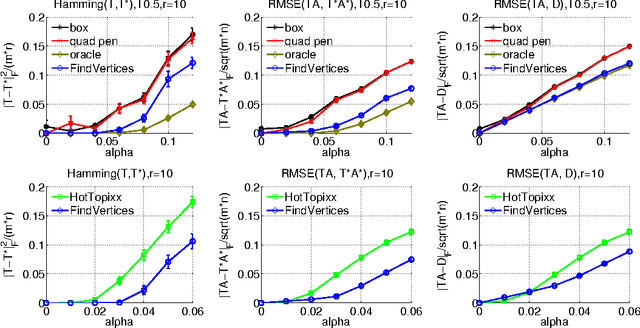
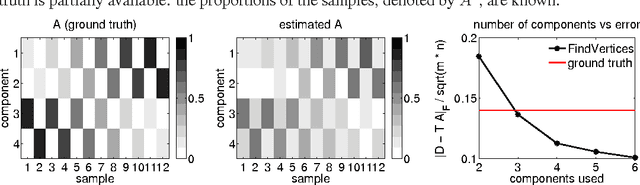
Motivated by an application in computational biology, we consider low-rank matrix factorization with $\{0,1\}$-constraints on one of the factors and optionally convex constraints on the second one. In addition to the non-convexity shared with other matrix factorization schemes, our problem is further complicated by a combinatorial constraint set of size $2^{m \cdot r}$, where $m$ is the dimension of the data points and $r$ the rank of the factorization. Despite apparent intractability, we provide - in the line of recent work on non-negative matrix factorization by Arora et al. (2012) - an algorithm that provably recovers the underlying factorization in the exact case with $O(m r 2^r + mnr + r^2 n)$ operations for $n$ datapoints. To obtain this result, we use theory around the Littlewood-Offord lemma from combinatorics.