 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImpact of Large Language Model Assistance on Patients Reading Clinical Notes: A Mixed-Methods Study
Jan 17, 2024



Patients derive numerous benefits from reading their clinical notes, including an increased sense of control over their health and improved understanding of their care plan. However, complex medical concepts and jargon within clinical notes hinder patient comprehension and may lead to anxiety. We developed a patient-facing tool to make clinical notes more readable, leveraging large language models (LLMs) to simplify, extract information from, and add context to notes. We prompt engineered GPT-4 to perform these augmentation tasks on real clinical notes donated by breast cancer survivors and synthetic notes generated by a clinician, a total of 12 notes with 3868 words. In June 2023, 200 female-identifying US-based participants were randomly assigned three clinical notes with varying levels of augmentations using our tool. Participants answered questions about each note, evaluating their understanding of follow-up actions and self-reported confidence. We found that augmentations were associated with a significant increase in action understanding score (0.63 $\pm$ 0.04 for select augmentations, compared to 0.54 $\pm$ 0.02 for the control) with p=0.002. In-depth interviews of self-identifying breast cancer patients (N=7) were also conducted via video conferencing. Augmentations, especially definitions, elicited positive responses among the seven participants, with some concerns about relying on LLMs. Augmentations were evaluated for errors by clinicians, and we found misleading errors occur, with errors more common in real donated notes than synthetic notes, illustrating the importance of carefully written clinical notes. Augmentations improve some but not all readability metrics. This work demonstrates the potential of LLMs to improve patients' experience with clinical notes at a lower burden to clinicians. However, having a human in the loop is important to correct potential model errors.
Conceptualizing Machine Learning for Dynamic Information Retrieval of Electronic Health Record Notes
Aug 09, 2023The large amount of time clinicians spend sifting through patient notes and documenting in electronic health records (EHRs) is a leading cause of clinician burnout. By proactively and dynamically retrieving relevant notes during the documentation process, we can reduce the effort required to find relevant patient history. In this work, we conceptualize the use of EHR audit logs for machine learning as a source of supervision of note relevance in a specific clinical context, at a particular point in time. Our evaluation focuses on the dynamic retrieval in the emergency department, a high acuity setting with unique patterns of information retrieval and note writing. We show that our methods can achieve an AUC of 0.963 for predicting which notes will be read in an individual note writing session. We additionally conduct a user study with several clinicians and find that our framework can help clinicians retrieve relevant information more efficiently. Demonstrating that our framework and methods can perform well in this demanding setting is a promising proof of concept that they will translate to other clinical settings and data modalities (e.g., labs, medications, imaging).
Machine Learning for Health symposium 2022 -- Extended Abstract track
Nov 28, 2022A collection of the extended abstracts that were presented at the 2nd Machine Learning for Health symposium (ML4H 2022), which was held both virtually and in person on November 28, 2022, in New Orleans, Louisiana, USA. Machine Learning for Health (ML4H) is a longstanding venue for research into machine learning for health, including both theoretical works and applied works. ML4H 2022 featured two submission tracks: a proceedings track, which encompassed full-length submissions of technically mature and rigorous work, and an extended abstract track, which would accept less mature, but innovative research for discussion. All the manuscripts submitted to ML4H Symposium underwent a double-blind peer-review process. Extended abstracts included in this collection describe innovative machine learning research focused on relevant problems in health and biomedicine.
TabLLM: Few-shot Classification of Tabular Data with Large Language Models
Oct 19, 2022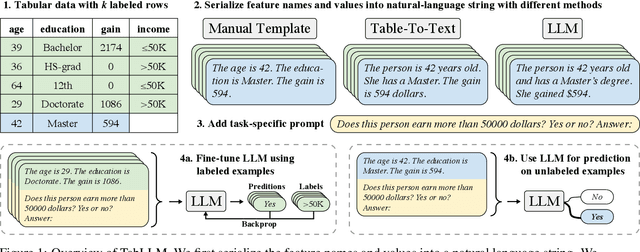
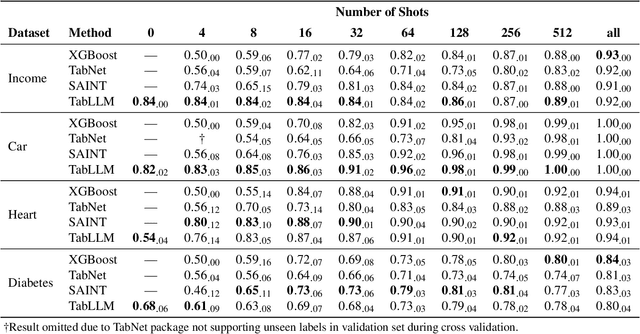
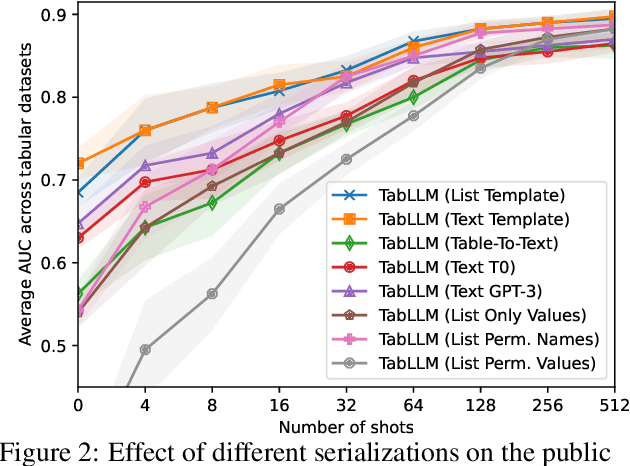
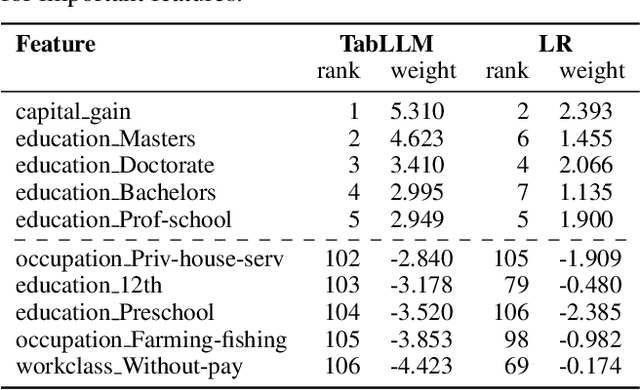
We study the application of large language models to zero-shot and few-shot classification of tabular data. We prompt the large language model with a serialization of the tabular data to a natural-language string, together with a short description of the classification problem. In the few-shot setting, we fine-tune the large language model using some labeled examples. We evaluate several serialization methods including templates, table-to-text models, and large language models. Despite its simplicity, we find that this technique outperforms prior deep-learning-based tabular classification methods on several benchmark datasets. In most cases, even zero-shot classification obtains non-trivial performance, illustrating the method's ability to exploit prior knowledge encoded in large language models. Unlike many deep learning methods for tabular datasets, this approach is also competitive with strong traditional baselines like gradient-boosted trees, especially in the very-few-shot setting.
Large Language Models are Zero-Shot Clinical Information Extractors
May 25, 2022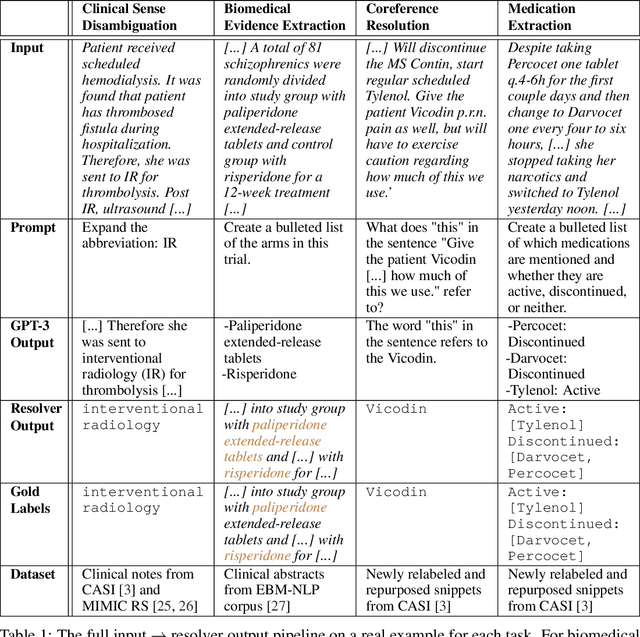


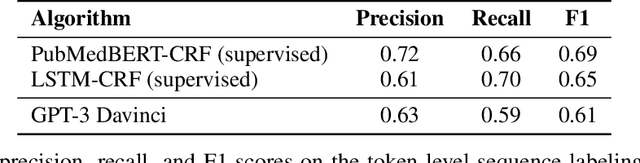
We show that large language models, such as GPT-3, perform well at zero-shot information extraction from clinical text despite not being trained specifically for the clinical domain. We present several examples showing how to use these models as tools for the diverse tasks of (i) concept disambiguation, (ii) evidence extraction, (iii) coreference resolution, and (iv) concept extraction, all on clinical text. The key to good performance is the use of simple task-specific programs that map from the language model outputs to the label space of the task. We refer to these programs as resolvers, a generalization of the verbalizer, which defines a mapping between output tokens and a discrete label space. We show in our examples that good resolvers share common components (e.g., "safety checks" that ensure the language model outputs faithfully match the input data), and that the common patterns across tasks make resolvers lightweight and easy to create. To better evaluate these systems, we also introduce two new datasets for benchmarking zero-shot clinical information extraction based on manual relabeling of the CASI dataset (Moon et al., 2014) with labels for new tasks. On the clinical extraction tasks we studied, the GPT-3 + resolver systems significantly outperform existing zero- and few-shot baselines.
Co-training Improves Prompt-based Learning for Large Language Models
Feb 02, 2022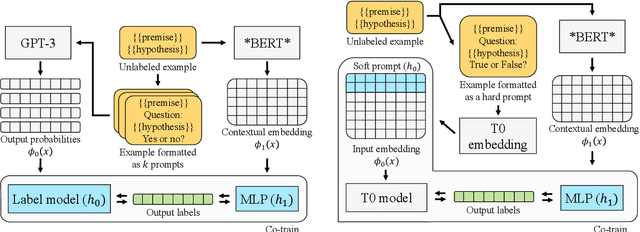
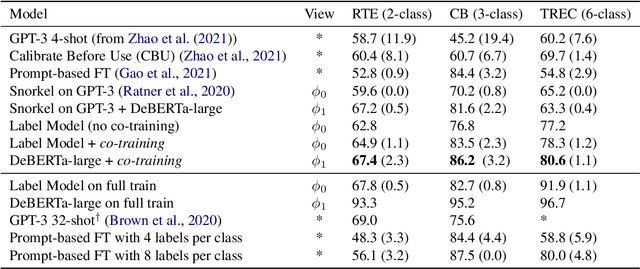
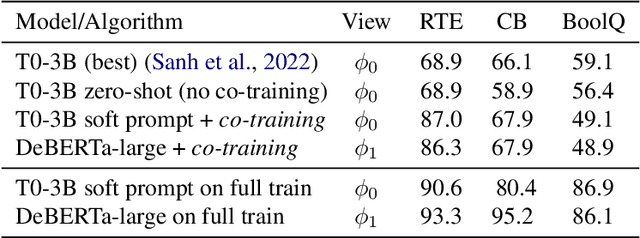
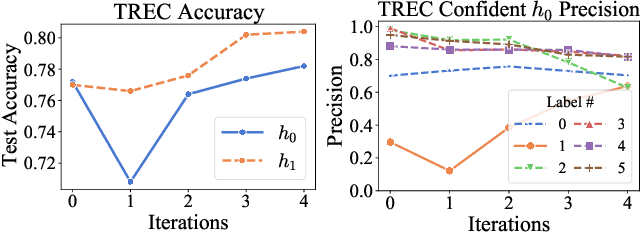
We demonstrate that co-training (Blum & Mitchell, 1998) can improve the performance of prompt-based learning by using unlabeled data. While prompting has emerged as a promising paradigm for few-shot and zero-shot learning, it is often brittle and requires much larger models compared to the standard supervised setup. We find that co-training makes it possible to improve the original prompt model and at the same time learn a smaller, downstream task-specific model. In the case where we only have partial access to a prompt model (e.g., output probabilities from GPT-3 (Brown et al., 2020)) we learn a calibration model over the prompt outputs. When we have full access to the prompt model's gradients but full finetuning remains prohibitively expensive (e.g., T0 (Sanh et al., 2021)), we learn a set of soft prompt continuous vectors to iteratively update the prompt model. We find that models trained in this manner can significantly improve performance on challenging datasets where there is currently a large gap between prompt-based learning and fully-supervised models.
Leveraging Time Irreversibility with Order-Contrastive Pre-training
Nov 04, 2021
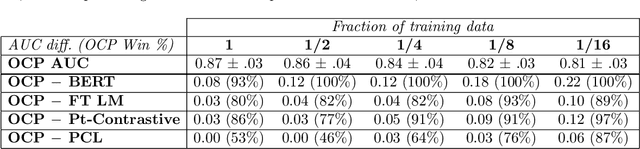
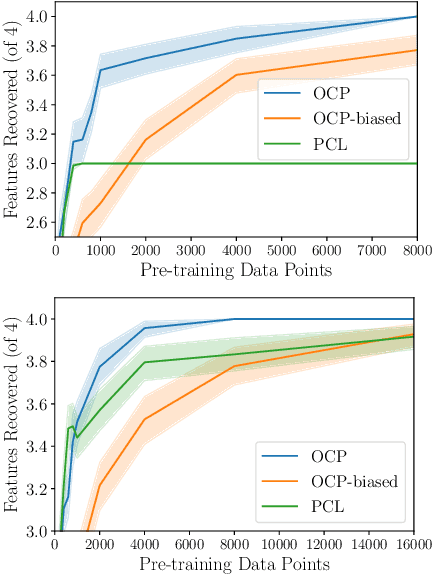
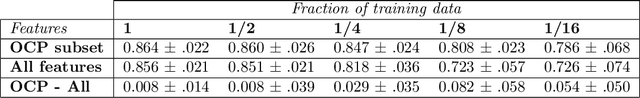
Label-scarce, high-dimensional domains such as healthcare present a challenge for modern machine learning techniques. To overcome the difficulties posed by a lack of labeled data, we explore an "order-contrastive" method for self-supervised pre-training on longitudinal data. We sample pairs of time segments, switch the order for half of them, and train a model to predict whether a given pair is in the correct order. Intuitively, the ordering task allows the model to attend to the least time-reversible features (for example, features that indicate progression of a chronic disease). The same features are often useful for downstream tasks of interest. To quantify this, we study a simple theoretical setting where we prove a finite-sample guarantee for the downstream error of a representation learned with order-contrastive pre-training. Empirically, in synthetic and longitudinal healthcare settings, we demonstrate the effectiveness of order-contrastive pre-training in the small-data regime over supervised learning and other self-supervised pre-training baselines. Our results indicate that pre-training methods designed for particular classes of distributions and downstream tasks can improve the performance of self-supervised learning.
PClean: Bayesian Data Cleaning at Scale with Domain-Specific Probabilistic Programming
Aug 07, 2020
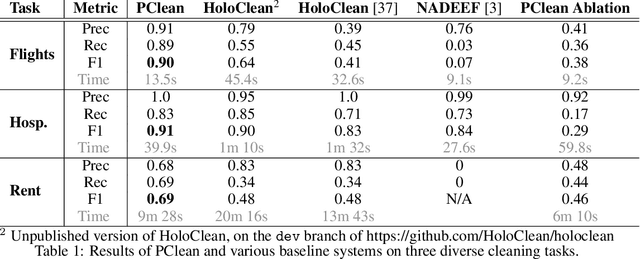

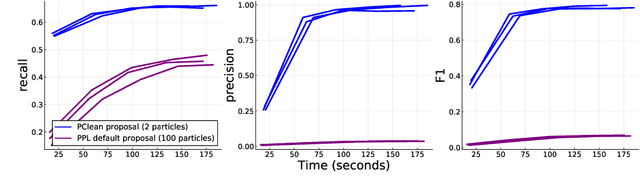
Data cleaning is naturally framed as probabilistic inference in a generative model, combining a prior distribution over ground-truth databases with a likelihood that models the noisy channel by which the data are filtered, corrupted, and joined to yield incomplete, dirty, and denormalized datasets. Based on this view, we present PClean, a unified generative modeling architecture for cleaning and normalizing dirty data in diverse domains. Given an unclean dataset and a probabilistic program encoding relevant domain knowledge, PClean learns a structured representation of the data as a relational database of interrelated objects, and uses this latent structure to impute missing values, identify duplicates, detect errors, and propose corrections in the original data table. PClean makes three modeling and inference contributions: (i) a domain-general non-parametric generative model of relational data, for inferring latent objects and their network of latent connections; (ii) a domain-specific probabilistic programming language, for encoding domain knowledge specific to each dataset being cleaned; and (iii) a domain-general inference engine that adapts to each PClean program by constructing data-driven proposals used in sequential Monte Carlo and particle Gibbs. We show empirically that short (< 50-line) PClean programs deliver higher accuracy than state-of-the-art data cleaning systems based on machine learning and weighted logic; that PClean's inference algorithm is faster than generic particle Gibbs inference for probabilistic programs; and that PClean scales to large real-world datasets with millions of rows.
Robust Benchmarking for Machine Learning of Clinical Entity Extraction
Jul 31, 2020
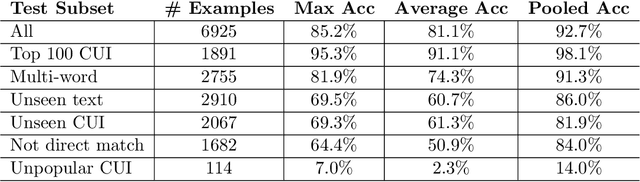
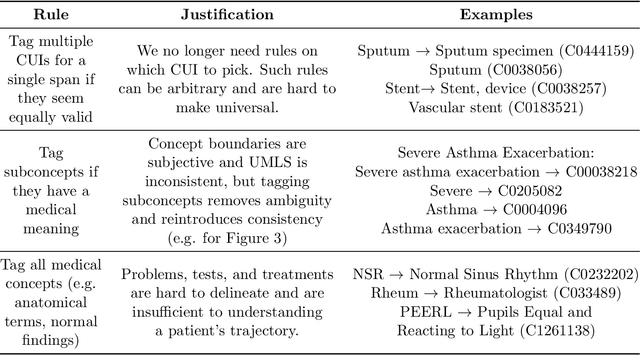
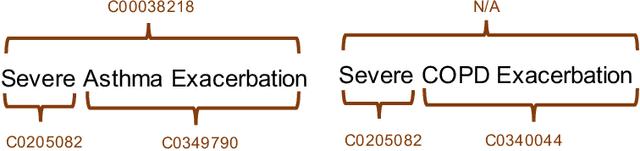
Clinical studies often require understanding elements of a patient's narrative that exist only in free text clinical notes. To transform notes into structured data for downstream use, these elements are commonly extracted and normalized to medical vocabularies. In this work, we audit the performance of and indicate areas of improvement for state-of-the-art systems. We find that high task accuracies for clinical entity normalization systems on the 2019 n2c2 Shared Task are misleading, and underlying performance is still brittle. Normalization accuracy is high for common concepts (95.3%), but much lower for concepts unseen in training data (69.3%). We demonstrate that current approaches are hindered in part by inconsistencies in medical vocabularies, limitations of existing labeling schemas, and narrow evaluation techniques. We reformulate the annotation framework for clinical entity extraction to factor in these issues to allow for robust end-to-end system benchmarking. We evaluate concordance of annotations from our new framework between two annotators and achieve a Jaccard similarity of 0.73 for entity recognition and an agreement of 0.83 for entity normalization. We propose a path forward to address the demonstrated need for the creation of a reference standard to spur method development in entity recognition and normalization.
Fast, Structured Clinical Documentation via Contextual Autocomplete
Jul 29, 2020

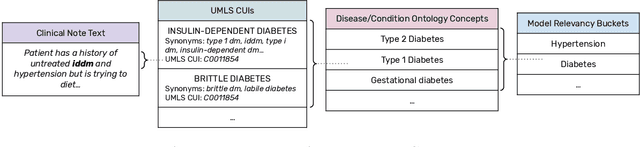

We present a system that uses a learned autocompletion mechanism to facilitate rapid creation of semi-structured clinical documentation. We dynamically suggest relevant clinical concepts as a doctor drafts a note by leveraging features from both unstructured and structured medical data. By constraining our architecture to shallow neural networks, we are able to make these suggestions in real time. Furthermore, as our algorithm is used to write a note, we can automatically annotate the documentation with clean labels of clinical concepts drawn from medical vocabularies, making notes more structured and readable for physicians, patients, and future algorithms. To our knowledge, this system is the only machine learning-based documentation utility for clinical notes deployed in a live hospital setting, and it reduces keystroke burden of clinical concepts by 67% in real environments.