 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUnderstood in Translation, Transformers for Domain Understanding
Dec 18, 2020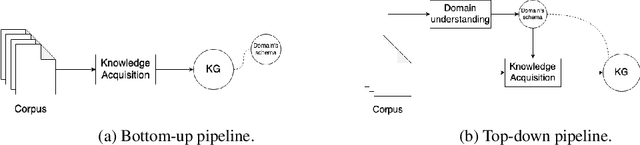
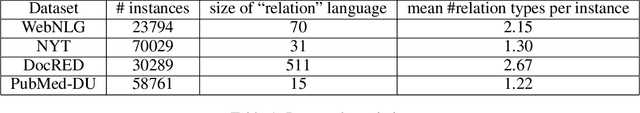
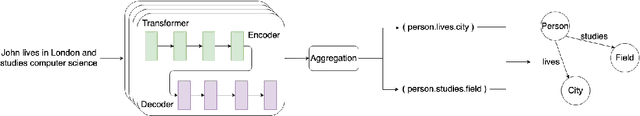
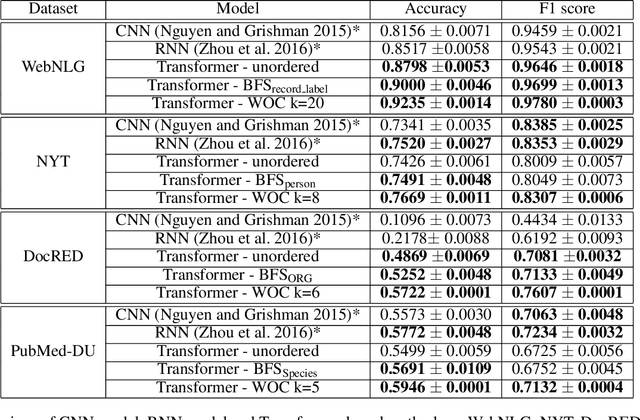
Knowledge acquisition is the essential first step of any Knowledge Graph (KG) application. This knowledge can be extracted from a given corpus (KG generation process) or specified from an existing KG (KG specification process). Focusing on domain specific solutions, knowledge acquisition is a labor intensive task usually orchestrated and supervised by subject matter experts. Specifically, the domain of interest is usually manually defined and then the needed generation or extraction tools are utilized to produce the KG. Herein, we propose a supervised machine learning method, based on Transformers, for domain definition of a corpus. We argue why such automated definition of the domain's structure is beneficial both in terms of construction time and quality of the generated graph. The proposed method is extensively validated on three public datasets (WebNLG, NYT and DocRED) by comparing it with two reference methods based on CNNs and RNNs models. The evaluation shows the efficiency of our model in this task. Focusing on scientific document understanding, we present a new health domain dataset based on publications extracted from PubMed and we successfully utilize our method on this. Lastly, we demonstrate how this work lays the foundation for fully automated and unsupervised KG generation.
Pre-training Protein Language Models with Label-Agnostic Binding Pairs Enhances Performance in Downstream Tasks
Dec 05, 2020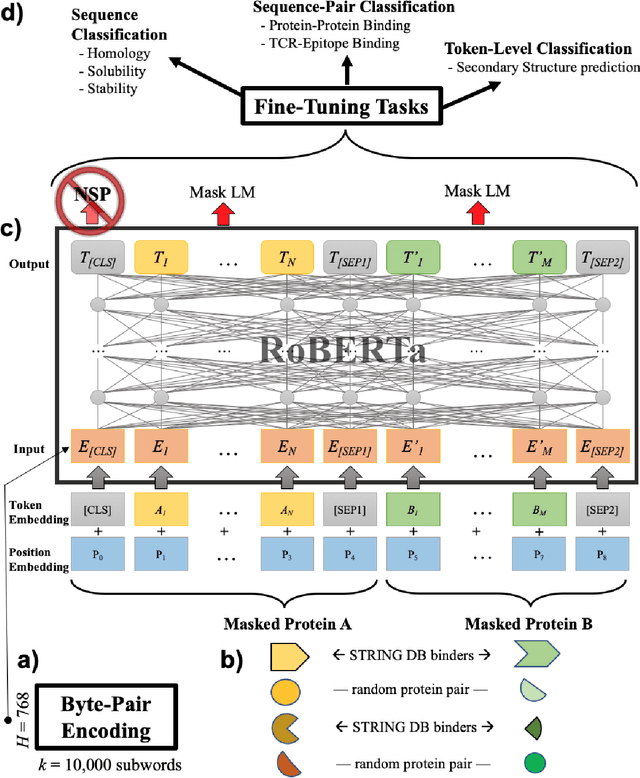
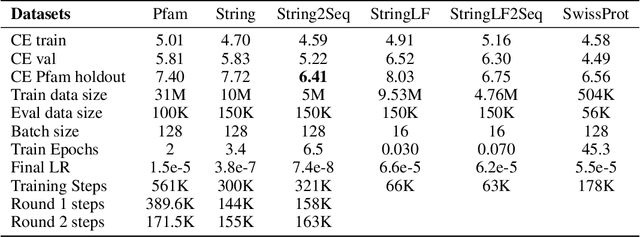

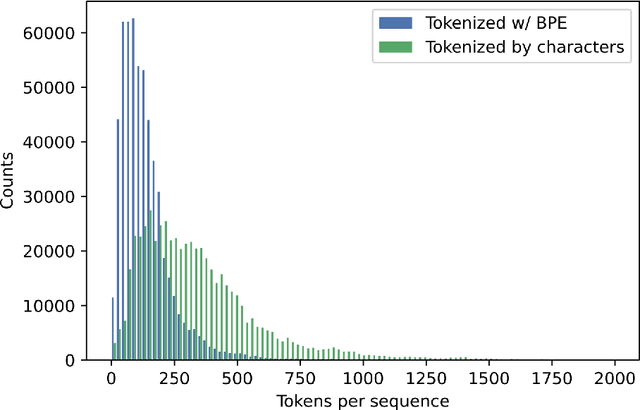
Less than 1% of protein sequences are structurally and functionally annotated. Natural Language Processing (NLP) community has recently embraced self-supervised learning as a powerful approach to learn representations from unlabeled text, in large part due to the attention-based context-aware Transformer models. In this work we present a modification to the RoBERTa model by inputting during pre-training a mixture of binding and non-binding protein sequences (from STRING database). However, the sequence pairs have no label to indicate their binding status, as the model relies solely on Masked Language Modeling (MLM) objective during pre-training. After fine-tuning, such approach surpasses models trained on single protein sequences for protein-protein binding prediction, TCR-epitope binding prediction, cellular-localization and remote homology classification tasks. We suggest that the Transformer's attention mechanism contributes to protein binding site discovery. Furthermore, we compress protein sequences by 64% with the Byte Pair Encoding (BPE) vocabulary consisting of 10K subwords, each around 3-4 amino acids long. Finally, to expand the model input space to even larger proteins and multi-protein assemblies, we pre-train Longformer models that support 2,048 tokens. Further work in token-level classification for secondary structure prediction is needed. Code available at: https://github.com/PaccMann/paccmann_proteomics
Hierarchical Pre-training for Sequence Labelling in Spoken Dialog
Oct 03, 2020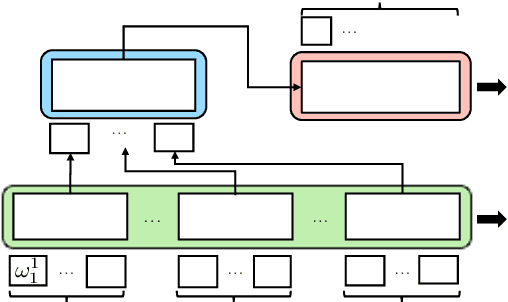
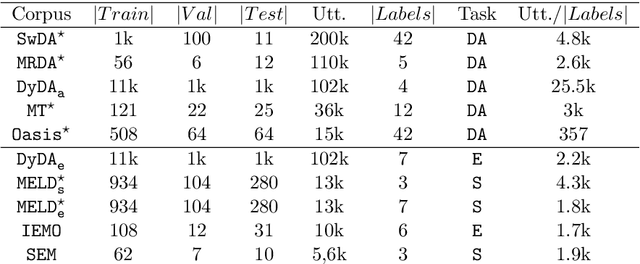
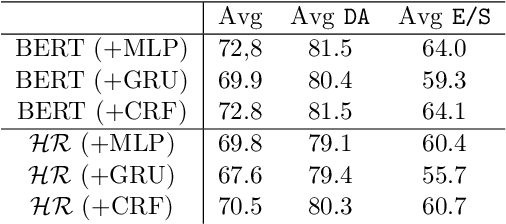
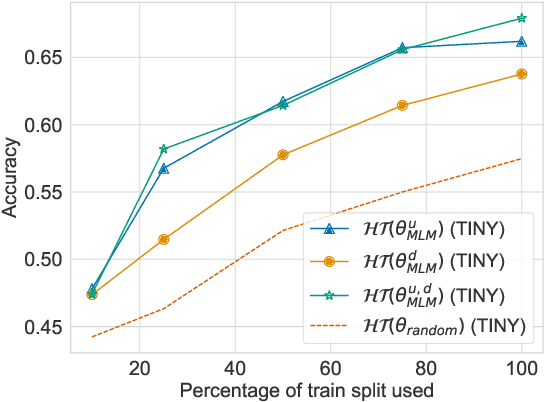
Sequence labelling tasks like Dialog Act and Emotion/Sentiment identification are a key component of spoken dialog systems. In this work, we propose a new approach to learn generic representations adapted to spoken dialog, which we evaluate on a new benchmark we call Sequence labellIng evaLuatIon benChmark fOr spoken laNguagE benchmark (\texttt{SILICONE}). \texttt{SILICONE} is model-agnostic and contains 10 different datasets of various sizes. We obtain our representations with a hierarchical encoder based on transformer architectures, for which we extend two well-known pre-training objectives. Pre-training is performed on OpenSubtitles: a large corpus of spoken dialog containing over $2.3$ billion of tokens. We demonstrate how hierarchical encoders achieve competitive results with consistently fewer parameters compared to state-of-the-art models and we show their importance for both pre-training and fine-tuning.
PaccMann$^{RL}$ on SARS-CoV-2: Designing antiviral candidates with conditional generative models
May 31, 2020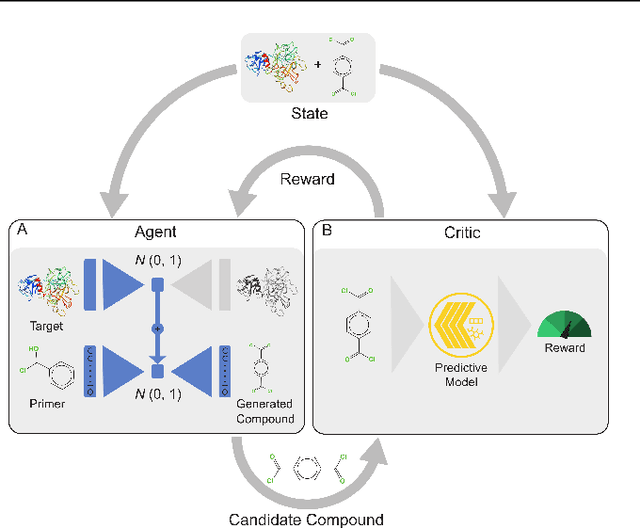

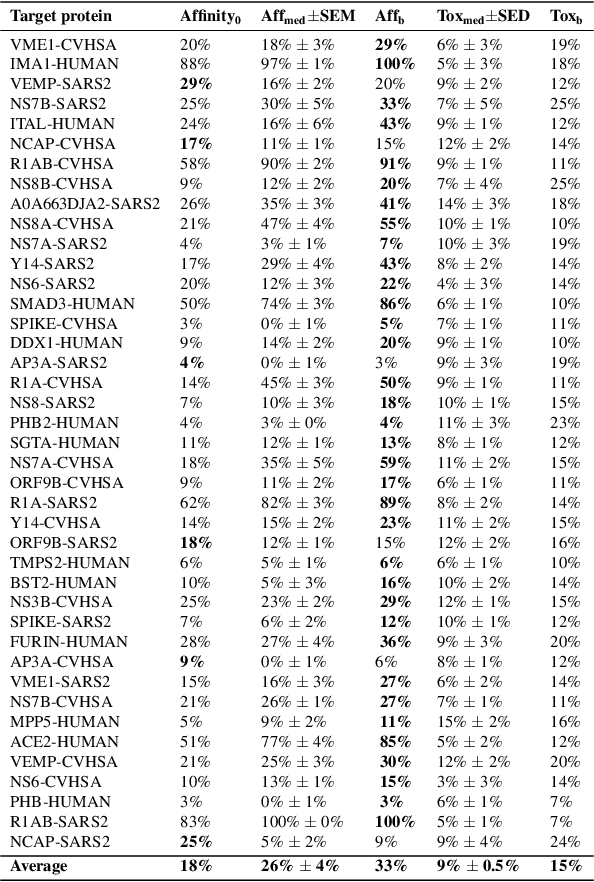
With the fast development of COVID-19 into a global pandemic, scientists around the globe are desperately searching for effective antiviral therapeutic agents. Bridging systems biology and drug discovery, we propose a deep learning framework for conditional de novo design of antiviral candidate drugs tailored against given protein targets. First, we train a multimodal ligand--protein binding affinity model on predicting affinities of antiviral compounds to target proteins and couple this model with pharmacological toxicity predictors. Exploiting this multi-objective as a reward function of a conditional molecular generator (consisting of two VAEs), we showcase a framework that navigates the chemical space toward regions with more antiviral molecules. Specifically, we explore a challenging setting of generating ligands against unseen protein targets by performing a leave-one-out-cross-validation on 41 SARS-CoV-2-related target proteins. Using deep RL, it is demonstrated that in 35 out of 41 cases, the generation is biased towards sampling more binding ligands, with an average increase of 83% comparing to an unbiased VAE. We present a case-study on a potential Envelope-protein inhibitor and perform a synthetic accessibility assessment of the best generated molecules is performed that resembles a viable roadmap towards a rapid in-vitro evaluation of potential SARS-CoV-2 inhibitors.
Guider l'attention dans les modeles de sequence a sequence pour la prediction des actes de dialogue
Feb 26, 2020
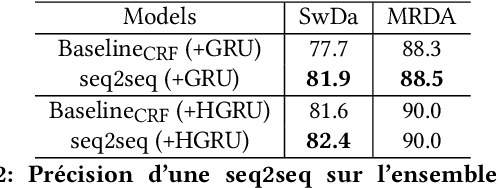
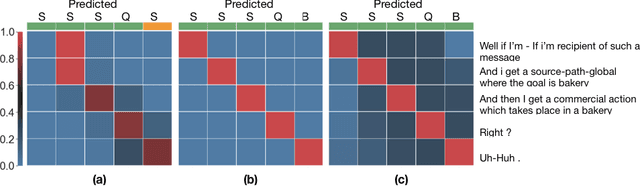

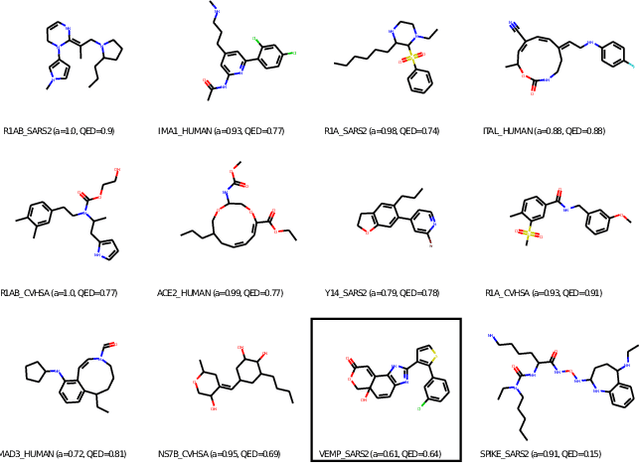
The task of predicting dialog acts (DA) based on conversational dialog is a key component in the development of conversational agents. Accurately predicting DAs requires a precise modeling of both the conversation and the global tag dependencies. We leverage seq2seq approaches widely adopted in Neural Machine Translation (NMT) to improve the modelling of tag sequentiality. Seq2seq models are known to learn complex global dependencies while currently proposed approaches using linear conditional random fields (CRF) only model local tag dependencies. In this work, we introduce a seq2seq model tailored for DA classification using: a hierarchical encoder, a novel guided attention mechanism and beam search applied to both training and inference. Compared to the state of the art our model does not require handcrafted features and is trained end-to-end. Furthermore, the proposed approach achieves an unmatched accuracy score of 85% on SwDA, and state-of-the-art accuracy score of 91.6% on MRDA.
* in French
Guiding attention in Sequence-to-sequence models for Dialogue Act prediction
Feb 26, 2020
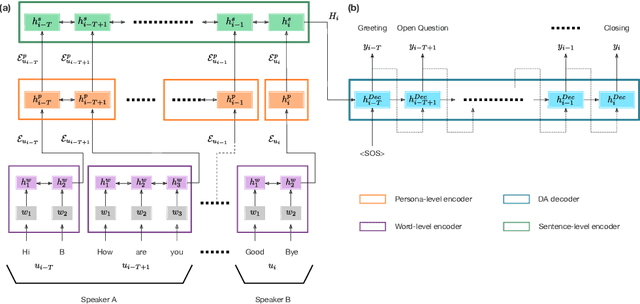
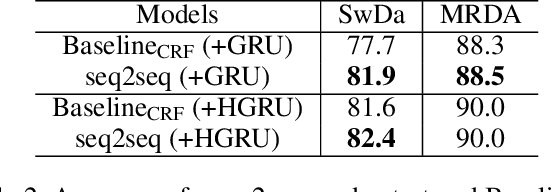
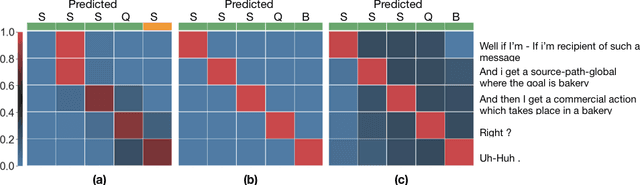
The task of predicting dialog acts (DA) based on conversational dialog is a key component in the development of conversational agents. Accurately predicting DAs requires a precise modeling of both the conversation and the global tag dependencies. We leverage seq2seq approaches widely adopted in Neural Machine Translation (NMT) to improve the modelling of tag sequentiality. Seq2seq models are known to learn complex global dependencies while currently proposed approaches using linear conditional random fields (CRF) only model local tag dependencies. In this work, we introduce a seq2seq model tailored for DA classification using: a hierarchical encoder, a novel guided attention mechanism and beam search applied to both training and inference. Compared to the state of the art our model does not require handcrafted features and is trained end-to-end. Furthermore, the proposed approach achieves an unmatched accuracy score of 85% on SwDA, and state-of-the-art accuracy score of 91.6% on MRDA.
Reinforcement learning-driven de-novo design of anticancer compounds conditioned on biomolecular profiles
Aug 29, 2019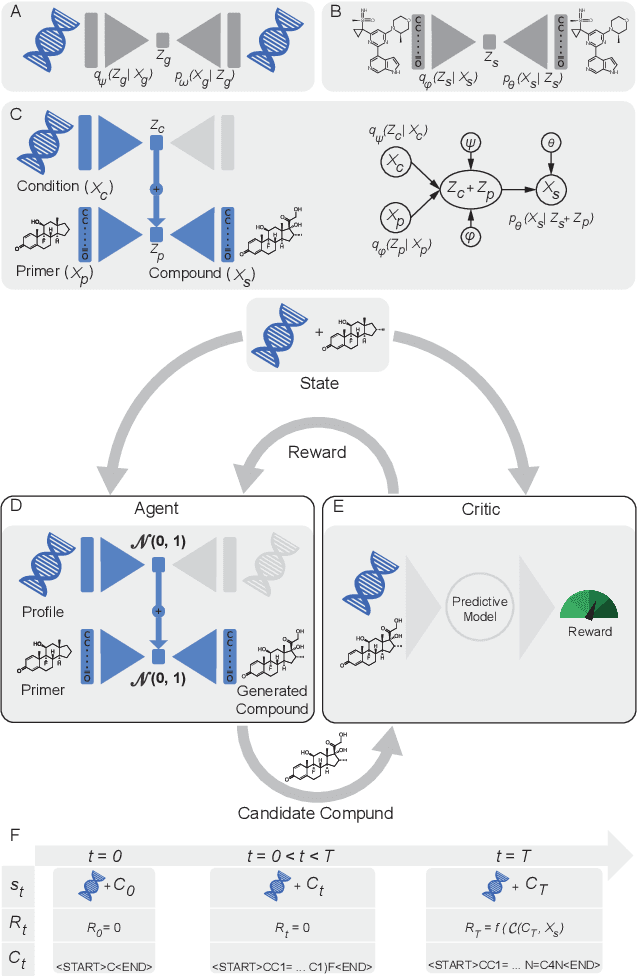
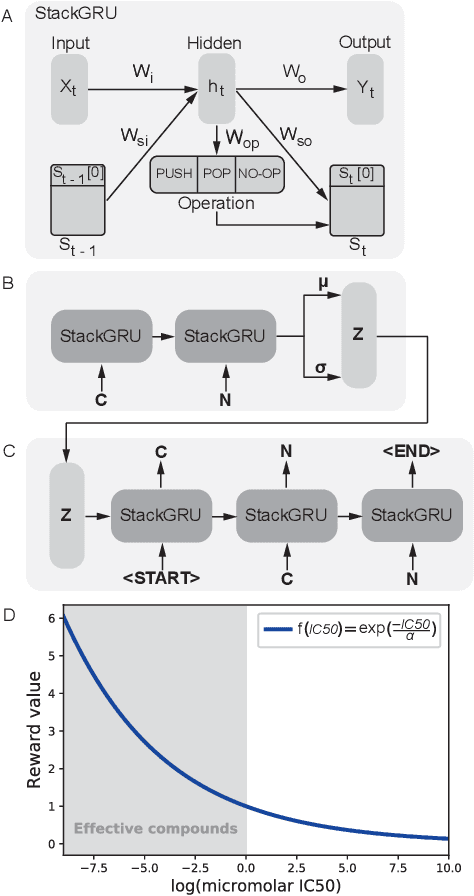
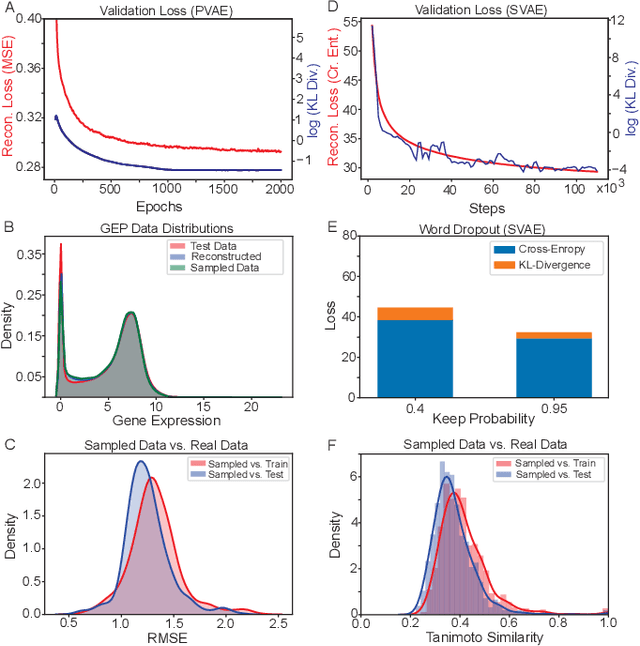
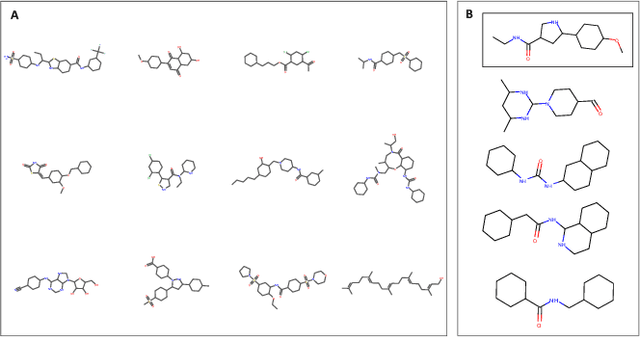
With the advent of deep generative models in computational chemistry, in silico anticancer drug design has undergone an unprecedented transformation. While state-of-the-art deep learning approaches have shown potential in generating compounds with desired chemical properties, they entirely overlook the genetic profile and properties of the target disease. In the case of cancer, this is problematic since it is a highly genetic disease in which the biomolecular profile of target cells determines the response to therapy. Here, we introduce the first deep generative model capable of generating anticancer compounds given a target biomolecular profile. Using a reinforcement learning framework, the transcriptomic profile of cancer cells is used as a context in which anticancer molecules are generated and optimized to obtain effective compounds for the given profile. Our molecule generator combines two pretrained variational autoencoders (VAEs) and a multimodal efficacy predictor - the first VAE generates transcriptomic profiles while the second conditional VAE generates novel molecular structures conditioned on the given transcriptomic profile. The efficacy predictor is used to optimize the generated molecules through a reward determined by the predicted IC50 drug sensitivity for the generated molecule and the target profile. We demonstrate how the molecule generation can be biased towards compounds with high inhibitory effect against individual cell lines or specific cancer sites. We verify our approach by investigating candidate drugs generated against specific cancer types and investigate their structural similarity to existing compounds with known efficacy against these cancer types. We envision our approach to transform in silico anticancer drug design by increasing success rates in lead compound discovery via leveraging the biomolecular characteristics of the disease.
An Information Extraction and Knowledge Graph Platform for Accelerating Biochemical Discoveries
Jul 19, 2019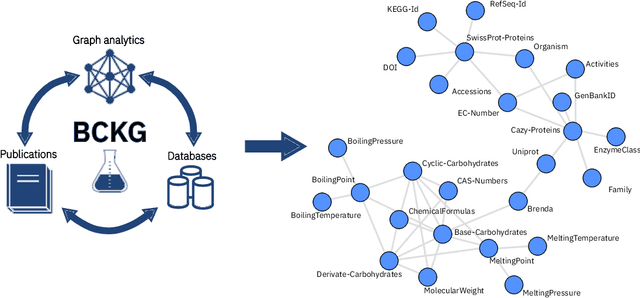
Information extraction and data mining in biochemical literature is a daunting task that demands resource-intensive computation and appropriate means to scale knowledge ingestion. Being able to leverage this immense source of technical information helps to drastically reduce costs and time to solution in multiple application fields from food safety to pharmaceutics. We present a scalable document ingestion system that integrates data from databases and publications (in PDF format) in a biochemistry knowledge graph (BCKG). The BCKG is a comprehensive source of knowledge that can be queried to retrieve known biochemical facts and to generate novel insights. After describing the knowledge ingestion framework, we showcase an application of our system in the field of carbohydrate enzymes. The BCKG represents a way to scale knowledge ingestion and automatically exploit prior knowledge to accelerate discovery in biochemical sciences.
Towards Explainable Anticancer Compound Sensitivity Prediction via Multimodal Attention-based Convolutional Encoders
May 22, 2019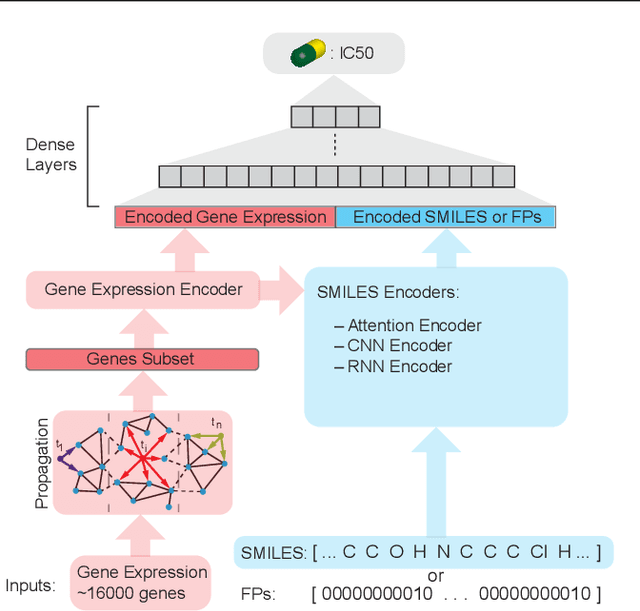
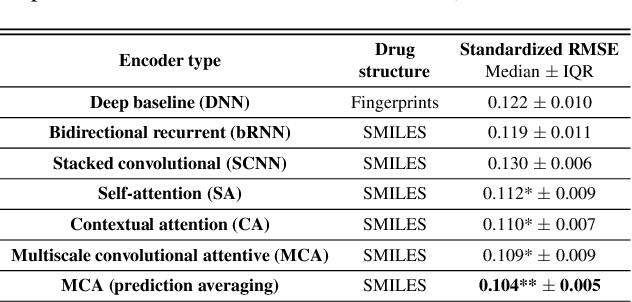
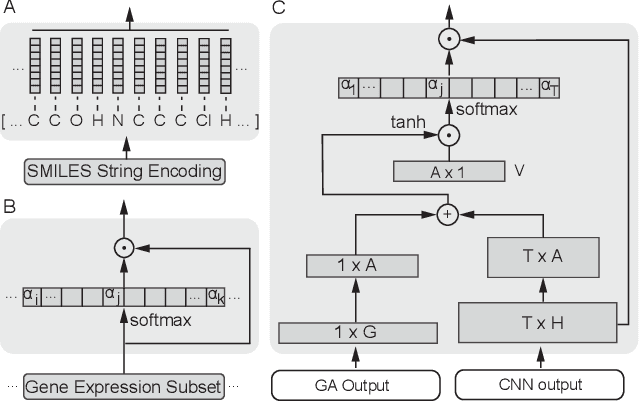
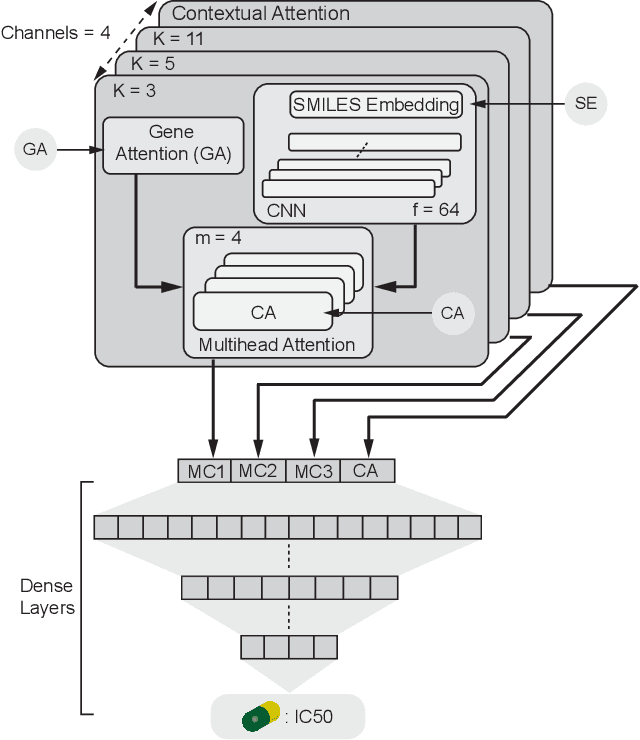
In line with recent advances in neural drug design and sensitivity prediction, we propose a novel architecture for interpretable prediction of anticancer compound sensitivity using a multimodal attention-based convolutional encoder. Our model is based on the three key pillars of drug sensitivity: compounds' structure in the form of a SMILES sequence, gene expression profiles of tumors and prior knowledge on intracellular interactions from protein-protein interaction networks. We demonstrate that our multiscale convolutional attention-based (MCA) encoder significantly outperforms a baseline model trained on Morgan fingerprints, a selection of encoders based on SMILES as well as previously reported state of the art for multimodal drug sensitivity prediction (R2 = 0.86 and RMSE = 0.89). Moreover, the explainability of our approach is demonstrated by a thorough analysis of the attention weights. We show that the attended genes significantly enrich apoptotic processes and that the drug attention is strongly correlated with a standard chemical structure similarity index. Finally, we report a case study of two receptor tyrosine kinase (RTK) inhibitors acting on a leukemia cell line, showcasing the ability of the model to focus on informative genes and submolecular regions of the two compounds. The demonstrated generalizability and the interpretability of our model testify its potential for in-silico prediction of anticancer compound efficacy on unseen cancer cells, positioning it as a valid solution for the development of personalized therapies as well as for the evaluation of candidate compounds in de novo drug design.
NeuNetS: An Automated Synthesis Engine for Neural Network Design
Jan 17, 2019
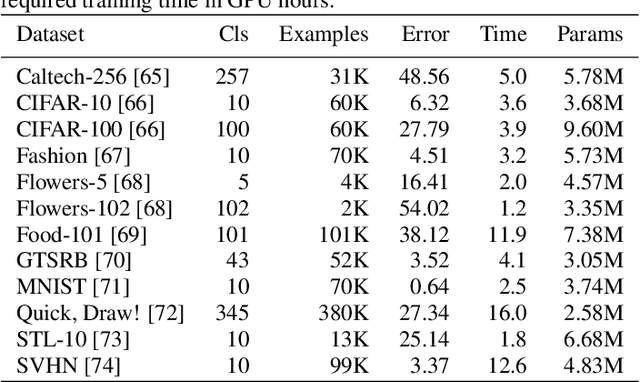
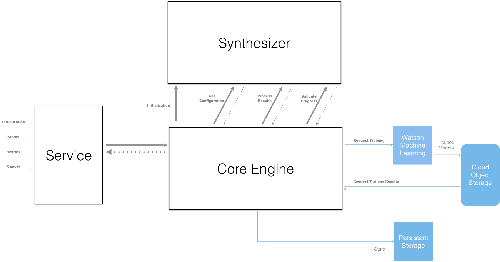
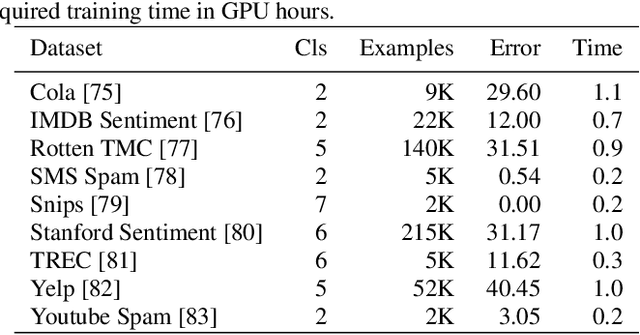
Application of neural networks to a vast variety of practical applications is transforming the way AI is applied in practice. Pre-trained neural network models available through APIs or capability to custom train pre-built neural network architectures with customer data has made the consumption of AI by developers much simpler and resulted in broad adoption of these complex AI models. While prebuilt network models exist for certain scenarios, to try and meet the constraints that are unique to each application, AI teams need to think about developing custom neural network architectures that can meet the tradeoff between accuracy and memory footprint to achieve the tight constraints of their unique use-cases. However, only a small proportion of data science teams have the skills and experience needed to create a neural network from scratch, and the demand far exceeds the supply. In this paper, we present NeuNetS : An automated Neural Network Synthesis engine for custom neural network design that is available as part of IBM's AI OpenScale's product. NeuNetS is available for both Text and Image domains and can build neural networks for specific tasks in a fraction of the time it takes today with human effort, and with accuracy similar to that of human-designed AI models.