 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-Scanner Canine Cutaneous Squamous Cell Carcinoma Histopathology Dataset
Jan 11, 2023

In histopathology, scanner-induced domain shifts are known to impede the performance of trained neural networks when tested on unseen data. Multi-domain pre-training or dedicated domain-generalization techniques can help to develop domain-agnostic algorithms. For this, multi-scanner datasets with a high variety of slide scanning systems are highly desirable. We present a publicly available multi-scanner dataset of canine cutaneous squamous cell carcinoma histopathology images, composed of 44 samples digitized with five slide scanners. This dataset provides local correspondences between images and thereby isolates the scanner-induced domain shift from other inherent, e.g. morphology-induced domain shifts. To highlight scanner differences, we present a detailed evaluation of color distributions, sharpness, and contrast of the individual scanner subsets. Additionally, to quantify the inherent scanner-induced domain shift, we train a tumor segmentation network on each scanner subset and evaluate the performance both in- and cross-domain. We achieve a class-averaged in-domain intersection over union coefficient of up to 0.86 and observe a cross-domain performance decrease of up to 0.38, which confirms the inherent domain shift of the presented dataset and its negative impact on the performance of deep neural networks.
Attention-based Multiple Instance Learning for Survival Prediction on Lung Cancer Tissue Microarrays
Dec 15, 2022

Attention-based multiple instance learning (AMIL) algorithms have proven to be successful in utilizing gigapixel whole-slide images (WSIs) for a variety of different computational pathology tasks such as outcome prediction and cancer subtyping problems. We extended an AMIL approach to the task of survival prediction by utilizing the classical Cox partial likelihood as a loss function, converting the AMIL model into a nonlinear proportional hazards model. We applied the model to tissue microarray (TMA) slides of 330 lung cancer patients. The results show that AMIL approaches can handle very small amounts of tissue from a TMA and reach similar C-index performance compared to established survival prediction methods trained with highly discriminative clinical factors such as age, cancer grade, and cancer stage
Deep Learning-Based Automatic Assessment of AgNOR-scores in Histopathology Images
Dec 15, 2022

Nucleolar organizer regions (NORs) are parts of the DNA that are involved in RNA transcription. Due to the silver affinity of associated proteins, argyrophilic NORs (AgNORs) can be visualized using silver-based staining. The average number of AgNORs per nucleus has been shown to be a prognostic factor for predicting the outcome of many tumors. Since manual detection of AgNORs is laborious, automation is of high interest. We present a deep learning-based pipeline for automatically determining the AgNOR-score from histopathological sections. An additional annotation experiment was conducted with six pathologists to provide an independent performance evaluation of our approach. Across all raters and images, we found a mean squared error of 0.054 between the AgNOR- scores of the experts and those of the model, indicating that our approach offers performance comparable to humans.
Deep learning-based Subtyping of Atypical and Normal Mitoses using a Hierarchical Anchor-Free Object Detector
Dec 12, 2022

Mitotic activity is key for the assessment of malignancy in many tumors. Moreover, it has been demonstrated that the proportion of abnormal mitosis to normal mitosis is of prognostic significance. Atypical mitotic figures (MF) can be identified morphologically as having segregation abnormalities of the chromatids. In this work, we perform, for the first time, automatic subtyping of mitotic figures into normal and atypical categories according to characteristic morphological appearances of the different phases of mitosis. Using the publicly available MIDOG21 and TUPAC16 breast cancer mitosis datasets, two experts blindly subtyped mitotic figures into five morphological categories. Further, we set up a state-of-the-art object detection pipeline extending the anchor-free FCOS approach with a gated hierarchical subclassification branch. Our labeling experiment indicated that subtyping of mitotic figures is a challenging task and prone to inter-rater disagreement, which we found in 24.89% of MF. Using the more diverse MIDOG21 dataset for training and TUPAC16 for testing, we reached a mean overall average precision score of 0.552, a ROC AUC score of 0.833 for atypical/normal MF and a mean class-averaged ROC-AUC score of 0.977 for discriminating the different phases of cells undergoing mitosis.
Mind the Gap: Scanner-induced domain shifts pose challenges for representation learning in histopathology
Nov 29, 2022



Computer-aided systems in histopathology are often challenged by various sources of domain shift that impact the performance of these algorithms considerably. We investigated the potential of using self-supervised pre-training to overcome scanner-induced domain shifts for the downstream task of tumor segmentation. For this, we present the Barlow Triplets to learn scanner-invariant representations from a multi-scanner dataset with local image correspondences. We show that self-supervised pre-training successfully aligned different scanner representations, which, interestingly only results in a limited benefit for our downstream task. We thereby provide insights into the influence of scanner characteristics for downstream applications and contribute to a better understanding of why established self-supervised methods have not yet shown the same success on histopathology data as they have for natural images.
Fast Text-Conditional Discrete Denoising on Vector-Quantized Latent Spaces
Nov 14, 2022



Conditional text-to-image generation has seen countless recent improvements in terms of quality, diversity and fidelity. Nevertheless, most state-of-the-art models require numerous inference steps to produce faithful generations, resulting in performance bottlenecks for end-user applications. In this paper we introduce Paella, a novel text-to-image model requiring less than 10 steps to sample high-fidelity images, using a speed-optimized architecture allowing to sample a single image in less than 500 ms, while having 573M parameters. The model operates on a compressed & quantized latent space, it is conditioned on CLIP embeddings and uses an improved sampling function over previous works. Aside from text-conditional image generation, our model is able to do latent space interpolation and image manipulations such as inpainting, outpainting, and structural editing. We release all of our code and pretrained models at https://github.com/dome272/Paella
Mitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022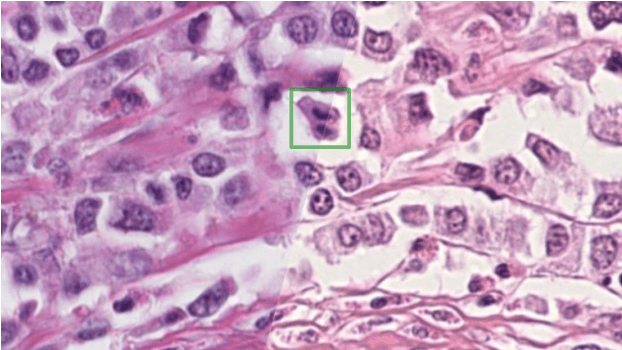
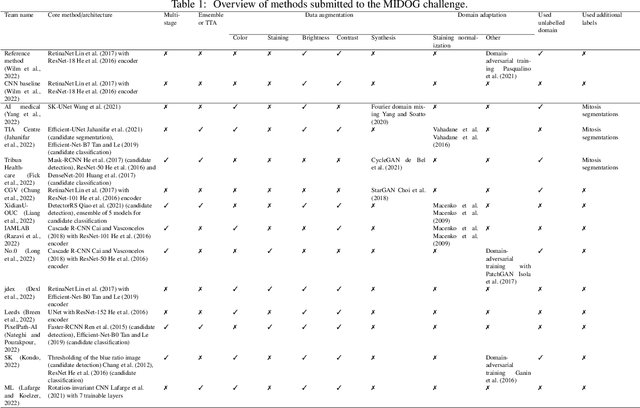
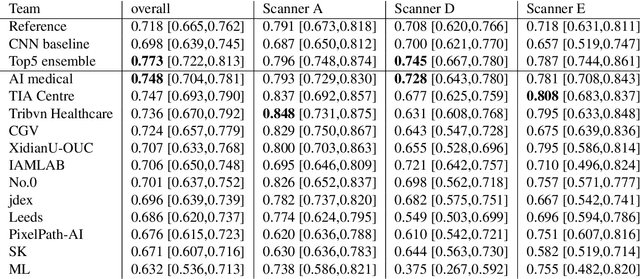
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.
Pan-Tumor CAnine cuTaneous Cancer Histology (CATCH) Dataset
Jan 27, 2022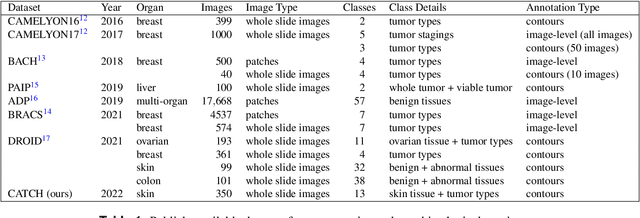
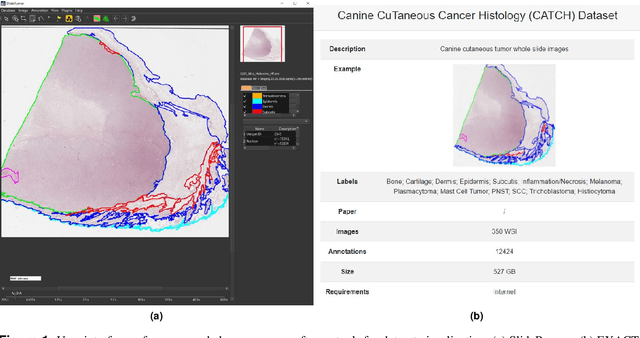
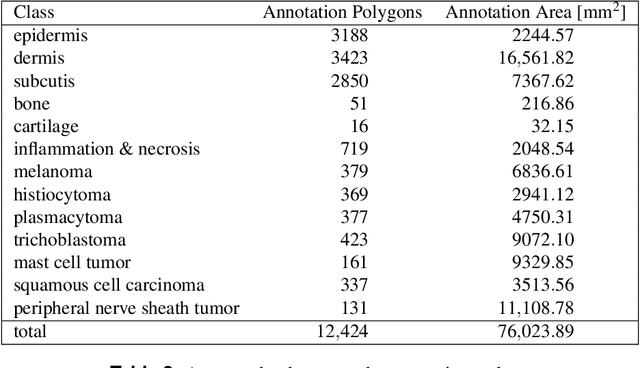

Due to morphological similarities, the differentiation of histologic sections of cutaneous tumors into individual subtypes can be challenging. Recently, deep learning-based approaches have proven their potential for supporting pathologists in this regard. However, many of these supervised algorithms require a large amount of annotated data for robust development. We present a publicly available dataset consisting of 350 whole slide images of seven different canine cutaneous tumors complemented by 12,424 polygon annotations for 13 histologic classes including seven cutaneous tumor subtypes. Regarding sample size and annotation extent, this exceeds most publicly available datasets which are oftentimes limited to the tumor area or merely provide patch-level annotations. We validated our model for tissue segmentation, achieving a class-averaged Jaccard coefficient of 0.7047, and 0.9044 for tumor in particular. For tumor subtype classification, we achieve a slide-level accuracy of 0.9857. Since canine cutaneous tumors possess various histologic homologies to human tumors, we believe that the added value of this dataset is not limited to veterinary pathology but extends to more general fields of application.
Domain Adversarial RetinaNet as a Reference Algorithm for the MItosis DOmain Generalization (MIDOG) Challenge
Aug 25, 2021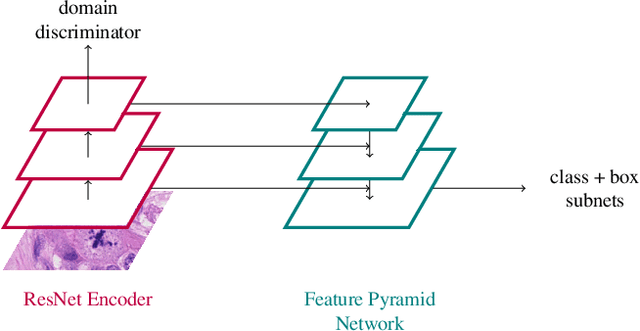
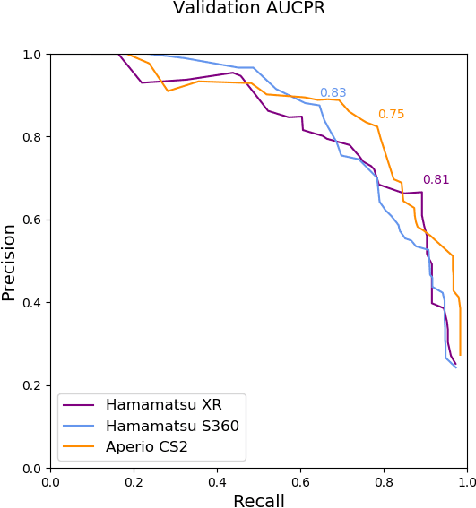
Assessing the Mitotic Count has a known high degree of intra- and inter-rater variability. Computer-aided systems have proven to decrease this variability and reduce labelling time. These systems, however, are generally highly dependent on their training domain and show poor applicability to unseen domains. In histopathology, these domain shifts can result from various sources, including different slide scanning systems used to digitize histologic samples. The MItosis DOmain Generalization challenge focuses on this specific domain shift for the task of mitotic figure detection. This work presents a mitotic figure detection algorithm developed as a baseline for the challenge, based on domain adversarial training. On the preliminary test set, the algorithm scores an F$_1$ score of 0.7514.
Inter-Species Cell Detection: Datasets on pulmonary hemosiderophages in equine, human and feline specimens
Aug 19, 2021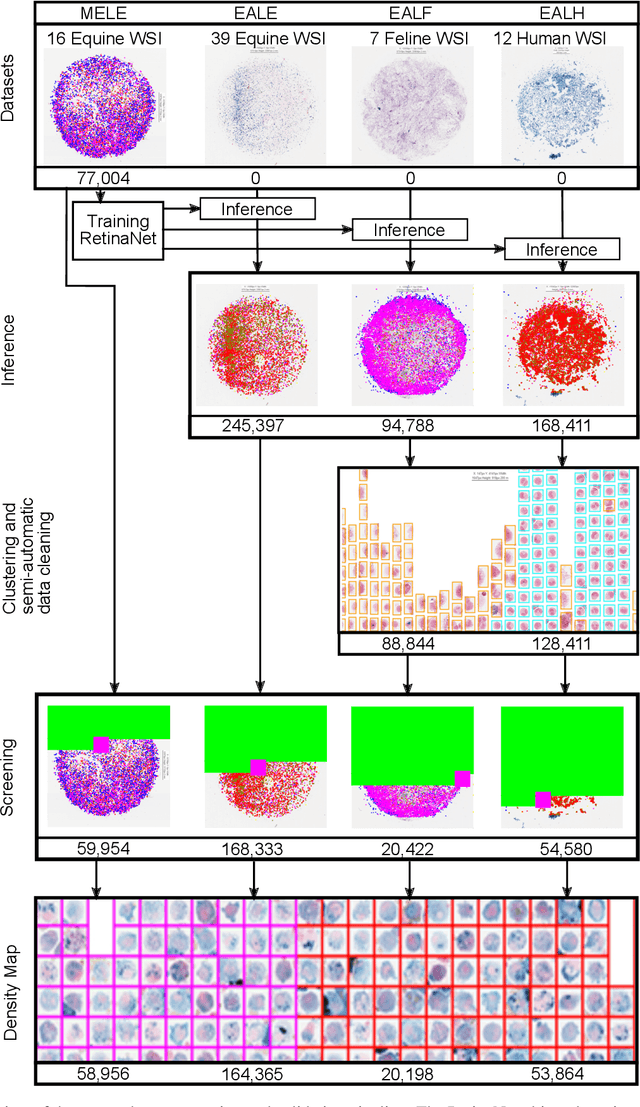
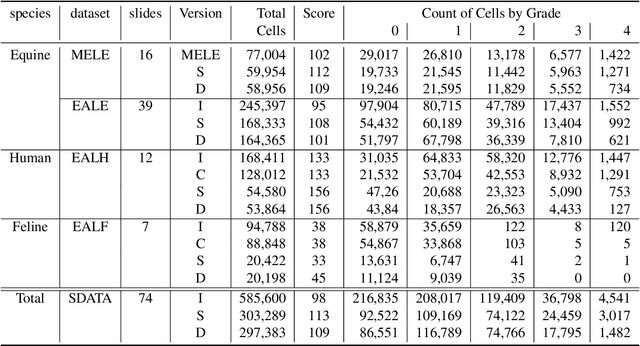
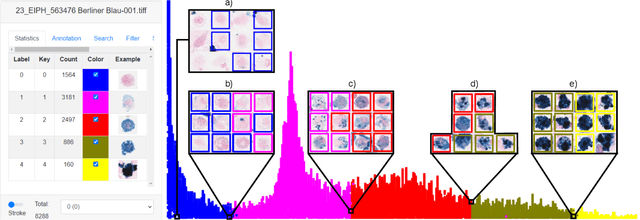
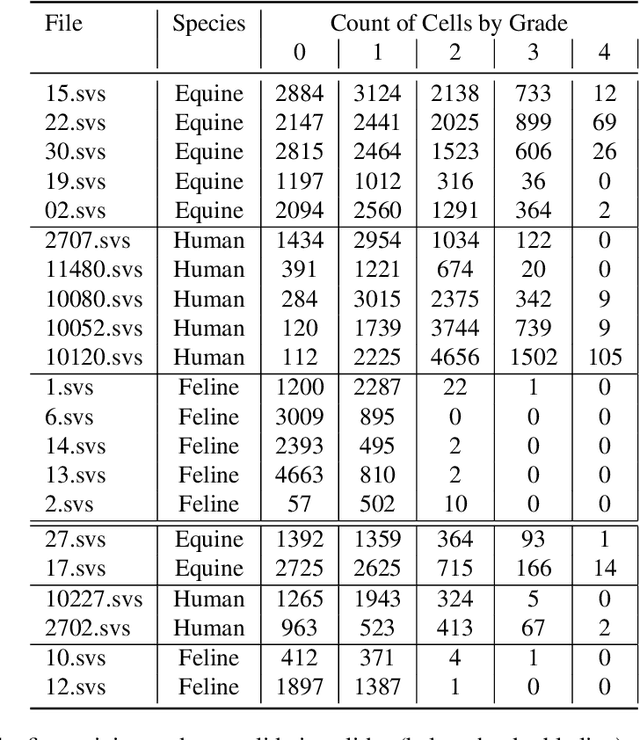
Pulmonary hemorrhage (P-Hem) occurs among multiple species and can have various causes. Cytology of bronchoalveolarlavage fluid (BALF) using a 5-tier scoring system of alveolar macrophages based on their hemosiderin content is considered the most sensitive diagnostic method. We introduce a novel, fully annotated multi-species P-Hem dataset which consists of 74 cytology whole slide images (WSIs) with equine, feline and human samples. To create this high-quality and high-quantity dataset, we developed an annotation pipeline combining human expertise with deep learning and data visualisation techniques. We applied a deep learning-based object detection approach trained on 17 expertly annotated equine WSIs, to the remaining 39 equine, 12 human and 7 feline WSIs. The resulting annotations were semi-automatically screened for errors on multiple types of specialised annotation maps and finally reviewed by a trained pathologists. Our dataset contains a total of 297,383 hemosiderophages classified into five grades. It is one of the largest publicly availableWSIs datasets with respect to the number of annotations, the scanned area and the number of species covered.