 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBreaking the Bottlenecks: Scalable Diffusion Models for 3D Molecular Generation
Jan 13, 2026Diffusion models have emerged as a powerful class of generative models for molecular design, capable of capturing complex structural distributions and achieving high fidelity in 3D molecule generation. However, their widespread use remains constrained by long sampling trajectories, stochastic variance in the reverse process, and limited structural awareness in denoising dynamics. The Directly Denoising Diffusion Model (DDDM) mitigates these inefficiencies by replacing stochastic reverse MCMC updates with deterministic denoising step, substantially reducing inference time. Yet, the theoretical underpinnings of such deterministic updates have remained opaque. In this work, we provide a principled reinterpretation of DDDM through the lens of the Reverse Transition Kernel (RTK) framework by Huang et al. 2024, unifying deterministic and stochastic diffusion under a shared probabilistic formalism. By expressing the DDDM reverse process as an approximate kernel operator, we show that the direct denoising process implicitly optimizes a structured transport map between noisy and clean samples. This perspective elucidates why deterministic denoising achieves efficient inference. Beyond theoretical clarity, this reframing resolves several long-standing bottlenecks in molecular diffusion. The RTK view ensures numerical stability by enforcing well-conditioned reverse kernels, improves sample consistency by eliminating stochastic variance, and enables scalable and symmetry-preserving denoisers that respect SE(3) equivariance. Empirically, we demonstrate that RTK-guided deterministic denoising achieves faster convergence and higher structural fidelity than stochastic diffusion models, while preserving chemical validity across GEOM-DRUGS dataset. Code, models, and datasets are publicly available in our project repository.
Classification in biological networks with hypergraphlet kernels
Mar 14, 2017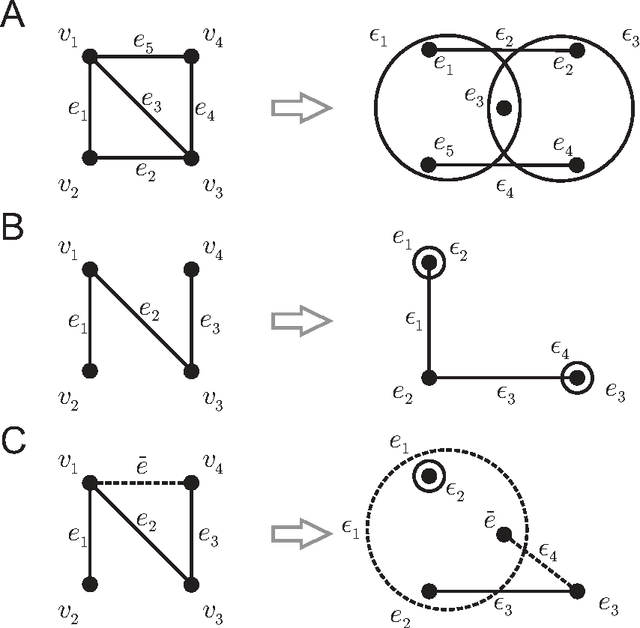
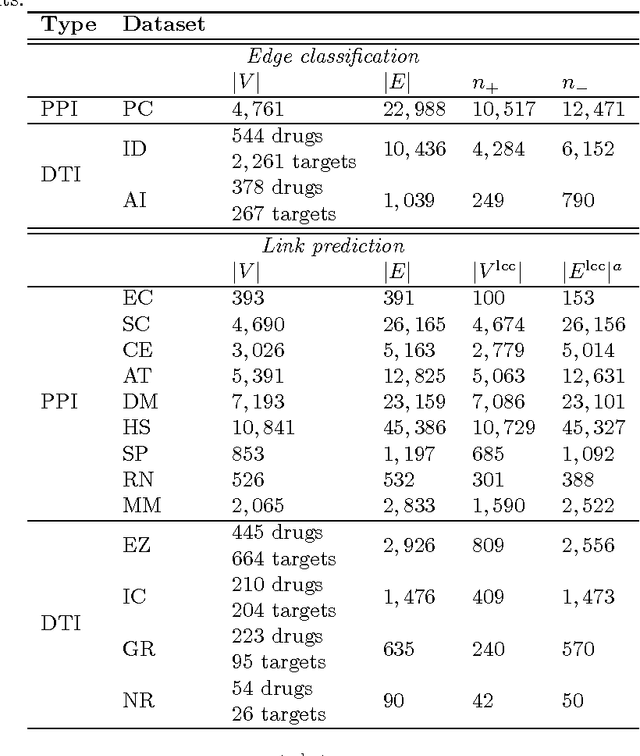
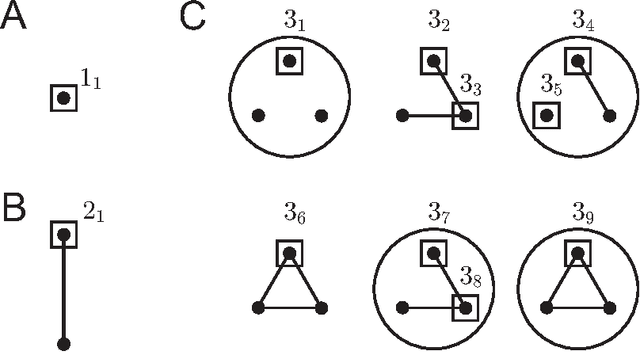
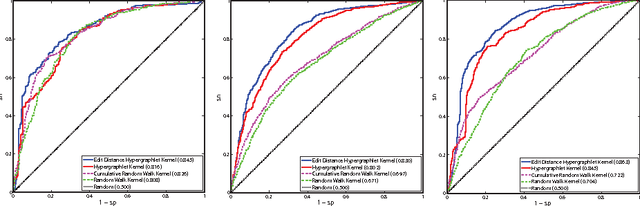
Biological and cellular systems are often modeled as graphs in which vertices represent objects of interest (genes, proteins, drugs) and edges represent relational ties among these objects (binds-to, interacts-with, regulates). This approach has been highly successful owing to the theory, methodology and software that support analysis and learning on graphs. Graphs, however, often suffer from information loss when modeling physical systems due to their inability to accurately represent multiobject relationships. Hypergraphs, a generalization of graphs, provide a framework to mitigate information loss and unify disparate graph-based methodologies. In this paper, we present a hypergraph-based approach for modeling physical systems and formulate vertex classification, edge classification and link prediction problems on (hyper)graphs as instances of vertex classification on (extended, dual) hypergraphs in a semi-supervised setting. We introduce a novel kernel method on vertex- and edge-labeled (colored) hypergraphs for analysis and learning. The method is based on exact and inexact (via hypergraph edit distances) enumeration of small simple hypergraphs, referred to as hypergraphlets, rooted at a vertex of interest. We extensively evaluate this method and show its potential use in a positive-unlabeled setting to estimate the number of missing and false positive links in protein-protein interaction networks.
Ultra High-Dimensional Nonlinear Feature Selection for Big Biological Data
Aug 14, 2016
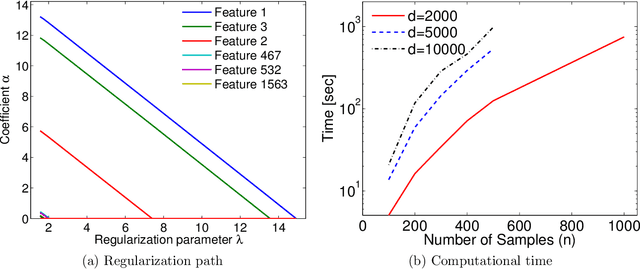
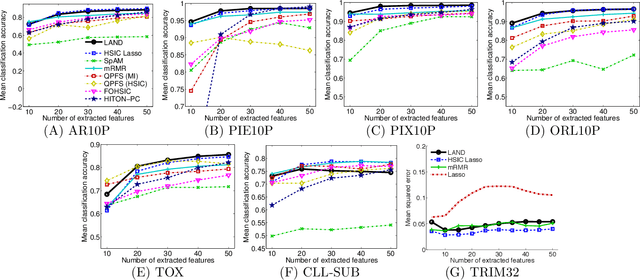
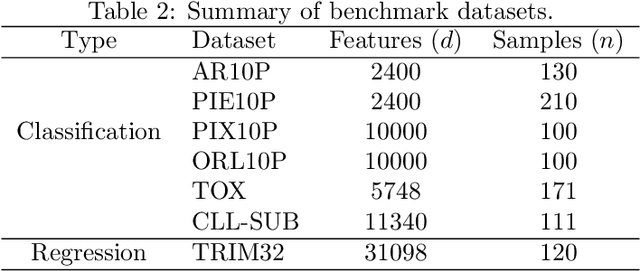
Machine learning methods are used to discover complex nonlinear relationships in biological and medical data. However, sophisticated learning models are computationally unfeasible for data with millions of features. Here we introduce the first feature selection method for nonlinear learning problems that can scale up to large, ultra-high dimensional biological data. More specifically, we scale up the novel Hilbert-Schmidt Independence Criterion Lasso (HSIC Lasso) to handle millions of features with tens of thousand samples. The proposed method is guaranteed to find an optimal subset of maximally predictive features with minimal redundancy, yielding higher predictive power and improved interpretability. Its effectiveness is demonstrated through applications to classify phenotypes based on module expression in human prostate cancer patients and to detect enzymes among protein structures. We achieve high accuracy with as few as 20 out of one million features --- a dimensionality reduction of 99.998%. Our algorithm can be implemented on commodity cloud computing platforms. The dramatic reduction of features may lead to the ubiquitous deployment of sophisticated prediction models in mobile health care applications.