 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecularIQ: Characterizing Chemical Reasoning Capabilities Through Symbolic Verification on Molecular Graphs
Jan 21, 2026A molecule's properties are fundamentally determined by its composition and structure encoded in its molecular graph. Thus, reasoning about molecular properties requires the ability to parse and understand the molecular graph. Large Language Models (LLMs) are increasingly applied to chemistry, tackling tasks such as molecular name conversion, captioning, text-guided generation, and property or reaction prediction. Most existing benchmarks emphasize general chemical knowledge, rely on literature or surrogate labels that risk leakage or bias, or reduce evaluation to multiple-choice questions. We introduce MolecularIQ, a molecular structure reasoning benchmark focused exclusively on symbolically verifiable tasks. MolecularIQ enables fine-grained evaluation of reasoning over molecular graphs and reveals capability patterns that localize model failures to specific tasks and molecular structures. This provides actionable insights into the strengths and limitations of current chemistry LLMs and guides the development of models that reason faithfully over molecular structure.
Measuring AI Progress in Drug Discovery: A Reproducible Leaderboard for the Tox21 Challenge
Nov 18, 2025Deep learning's rise since the early 2010s has transformed fields like computer vision and natural language processing and strongly influenced biomedical research. For drug discovery specifically, a key inflection - akin to vision's "ImageNet moment" - arrived in 2015, when deep neural networks surpassed traditional approaches on the Tox21 Data Challenge. This milestone accelerated the adoption of deep learning across the pharmaceutical industry, and today most major companies have integrated these methods into their research pipelines. After the Tox21 Challenge concluded, its dataset was included in several established benchmarks, such as MoleculeNet and the Open Graph Benchmark. However, during these integrations, the dataset was altered and labels were imputed or manufactured, resulting in a loss of comparability across studies. Consequently, the extent to which bioactivity and toxicity prediction methods have improved over the past decade remains unclear. To this end, we introduce a reproducible leaderboard, hosted on Hugging Face with the original Tox21 Challenge dataset, together with a set of baseline and representative methods. The current version of the leaderboard indicates that the original Tox21 winner - the ensemble-based DeepTox method - and the descriptor-based self-normalizing neural networks introduced in 2017, continue to perform competitively and rank among the top methods for toxicity prediction, leaving it unclear whether substantial progress in toxicity prediction has been achieved over the past decade. As part of this work, we make all baselines and evaluated models publicly accessible for inference via standardized API calls to Hugging Face Spaces.
Bio-xLSTM: Generative modeling, representation and in-context learning of biological and chemical sequences
Nov 06, 2024



Language models for biological and chemical sequences enable crucial applications such as drug discovery, protein engineering, and precision medicine. Currently, these language models are predominantly based on Transformer architectures. While Transformers have yielded impressive results, their quadratic runtime dependency on the sequence length complicates their use for long genomic sequences and in-context learning on proteins and chemical sequences. Recently, the recurrent xLSTM architecture has been shown to perform favorably compared to Transformers and modern state-space model (SSM) architectures in the natural language domain. Similar to SSMs, xLSTMs have a linear runtime dependency on the sequence length and allow for constant-memory decoding at inference time, which makes them prime candidates for modeling long-range dependencies in biological and chemical sequences. In this work, we tailor xLSTM towards these domains and propose a suite of architectural variants called Bio-xLSTM. Extensive experiments in three large domains, genomics, proteins, and chemistry, were performed to assess xLSTM's ability to model biological and chemical sequences. The results show that models based on Bio-xLSTM a) can serve as proficient generative models for DNA, protein, and chemical sequences, b) learn rich representations for those modalities, and c) can perform in-context learning for proteins and small molecules.
Large-scale ligand-based virtual screening for SARS-CoV-2 inhibitors using deep neural networks
Apr 03, 2020
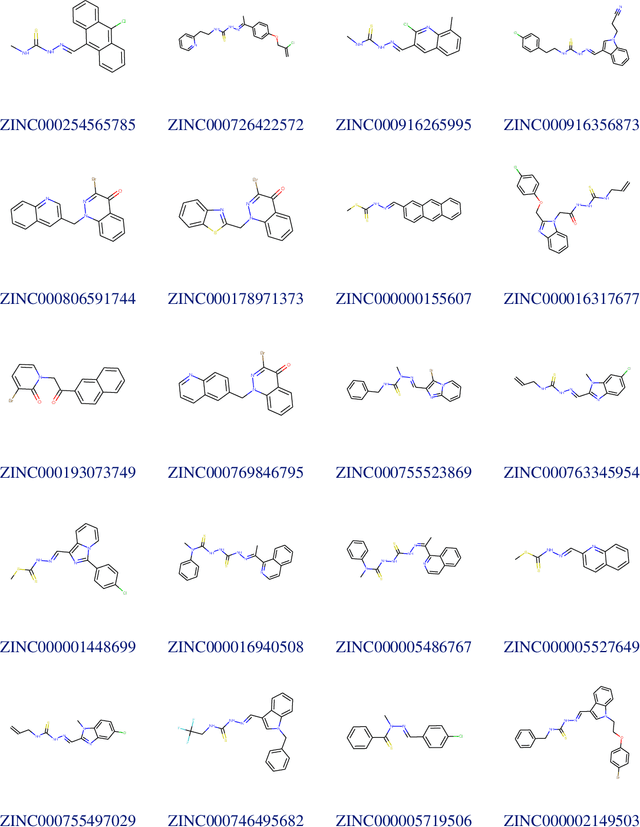

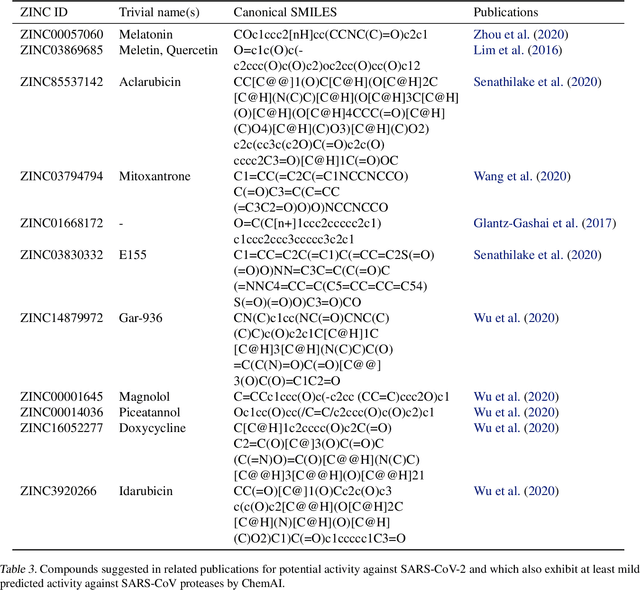
Due to the current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic, there is an urgent need for novel therapies and drugs. We conducted a large-scale virtual screening for small molecules that are potential CoV-2 inhibitors. To this end, we utilized "ChemAI", a deep neural network trained on more than 220M data points across 3.6M molecules from three public drug-discovery databases. With ChemAI, we screened and ranked one billion molecules from the ZINC database for favourable effects against CoV-2. We then reduced the result to the 30,000 top-ranked compounds, which are readily accessible and purchasable via the ZINC database. Additionally, we screened the DrugBank using ChemAI to allow for drug repurposing, which would be a fast way towards a therapy. We provide these top-ranked compounds of ZINC and DrugBank as a library for further screening with bioassays at https://github.com/ml-jku/sars-cov-inhibitors-chemai.