 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeStochastic Learning for Sparse Discrete Markov Random Fields with Controlled Gradient Approximation Error
May 12, 2020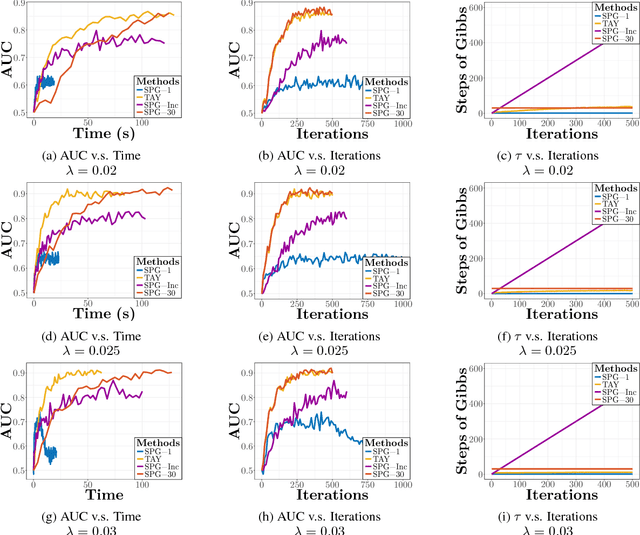
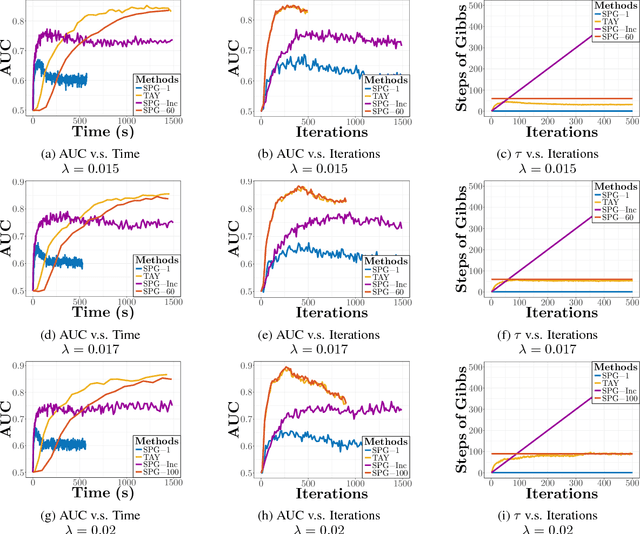
We study the $L_1$-regularized maximum likelihood estimator/estimation (MLE) problem for discrete Markov random fields (MRFs), where efficient and scalable learning requires both sparse regularization and approximate inference. To address these challenges, we consider a stochastic learning framework called stochastic proximal gradient (SPG; Honorio 2012a, Atchade et al. 2014,Miasojedow and Rejchel 2016). SPG is an inexact proximal gradient algorithm [Schmidtet al., 2011], whose inexactness stems from the stochastic oracle (Gibbs sampling) for gradient approximation - exact gradient evaluation is infeasible in general due to the NP-hard inference problem for discrete MRFs [Koller and Friedman, 2009]. Theoretically, we provide novel verifiable bounds to inspect and control the quality of gradient approximation. Empirically, we propose the tighten asymptotically (TAY) learning strategy based on the verifiable bounds to boost the performance of SPG.
CAUSE: Learning Granger Causality from Event Sequences using Attribution Methods
Feb 18, 2020
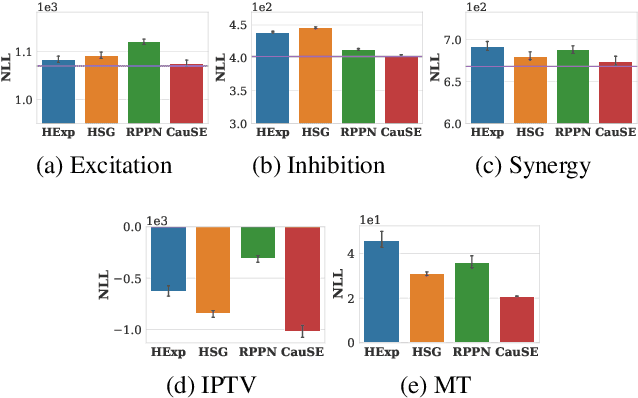
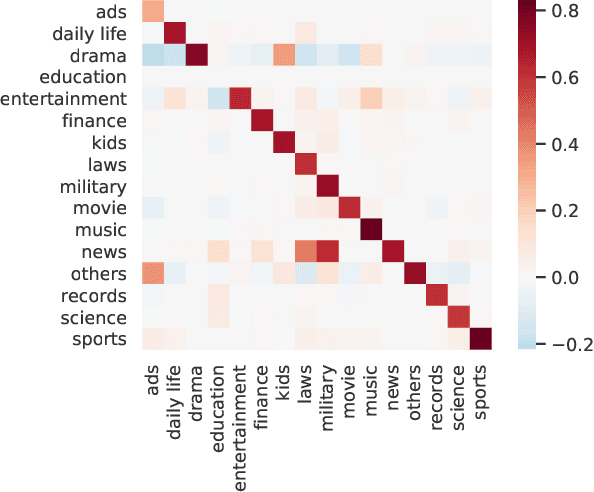
We study the problem of learning Granger causality between event types from asynchronous, interdependent, multi-type event sequences. Existing work suffers from either limited model flexibility or poor model explainability and thus fails to uncover Granger causality across a wide variety of event sequences with diverse event interdependency. To address these weaknesses, we propose CAUSE (Causality from AttribUtions on Sequence of Events), a novel framework for the studied task. The key idea of CAUSE is to first implicitly capture the underlying event interdependency by fitting a neural point process, and then extract from the process a Granger causality statistic using an axiomatic attribution method. Across multiple datasets riddled with diverse event interdependency, we demonstrate that CAUSE achieves superior performance on correctly inferring the inter-type Granger causality over a range of state-of-the-art methods.
AutoBlock: A Hands-off Blocking Framework for Entity Matching
Dec 07, 2019
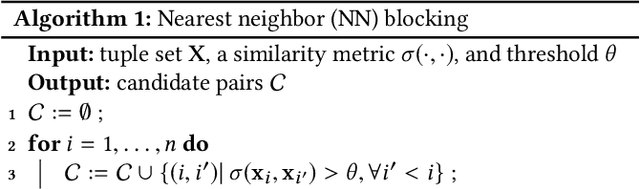
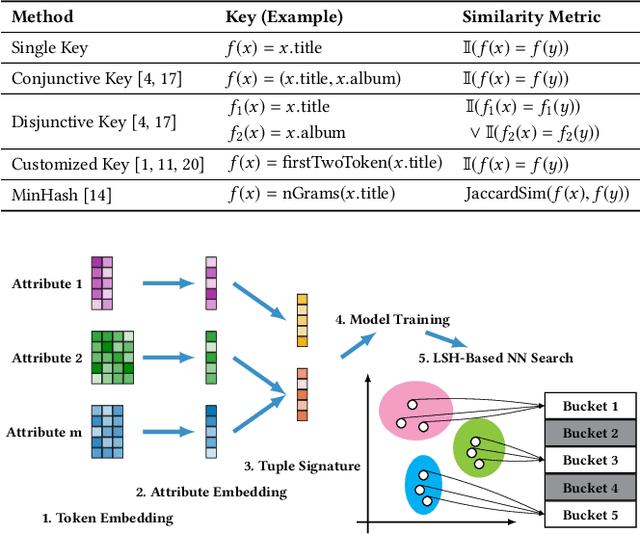

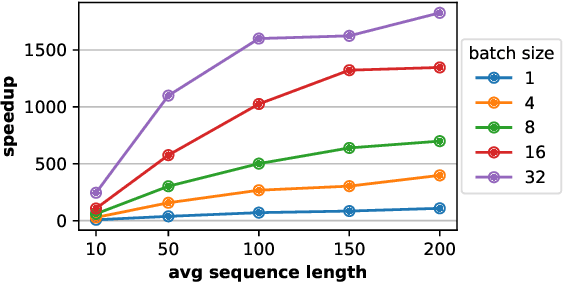
Entity matching seeks to identify data records over one or multiple data sources that refer to the same real-world entity. Virtually every entity matching task on large datasets requires blocking, a step that reduces the number of record pairs to be matched. However, most of the traditional blocking methods are learning-free and key-based, and their successes are largely built on laborious human effort in cleaning data and designing blocking keys. In this paper, we propose AutoBlock, a novel hands-off blocking framework for entity matching, based on similarity-preserving representation learning and nearest neighbor search. Our contributions include: (a) Automation: AutoBlock frees users from laborious data cleaning and blocking key tuning. (b) Scalability: AutoBlock has a sub-quadratic total time complexity and can be easily deployed for millions of records. (c) Effectiveness: AutoBlock outperforms a wide range of competitive baselines on multiple large-scale, real-world datasets, especially when datasets are dirty and/or unstructured.
Predicting Drug-Drug Interactions from Molecular Structure Images
Nov 14, 2019
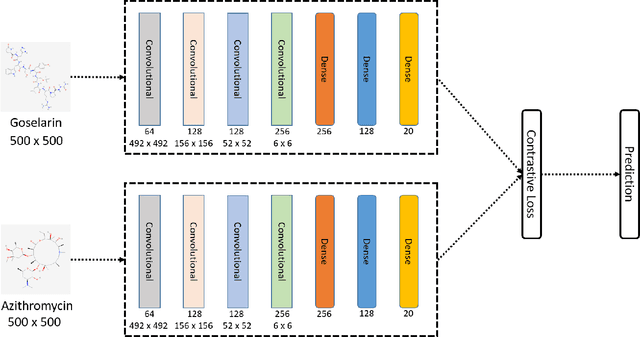
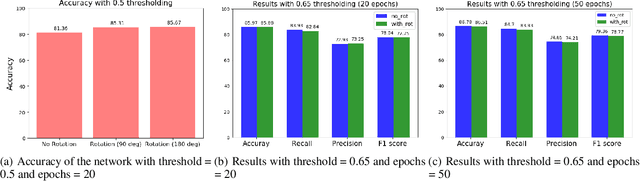
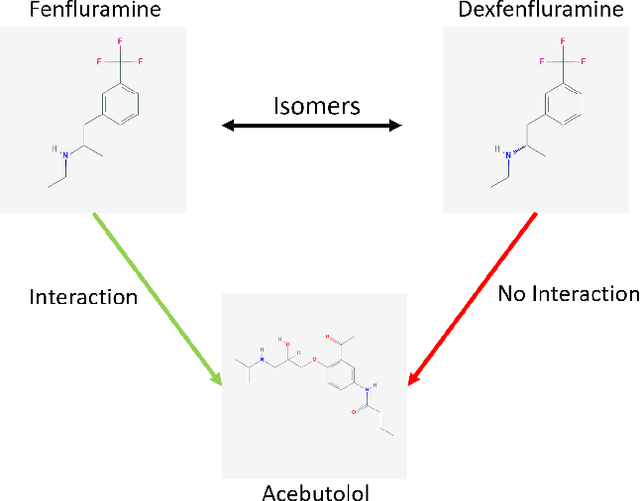
Predicting and discovering drug-drug interactions (DDIs) is an important problem and has been studied extensively both from medical and machine learning point of view. Almost all of the machine learning approaches have focused on text data or textual representation of the structural data of drugs. We present the first work that uses drug structure images as the input and utilizes a Siamese convolutional network architecture to predict DDIs.
High-Throughput Machine Learning from Electronic Health Records
Jul 03, 2019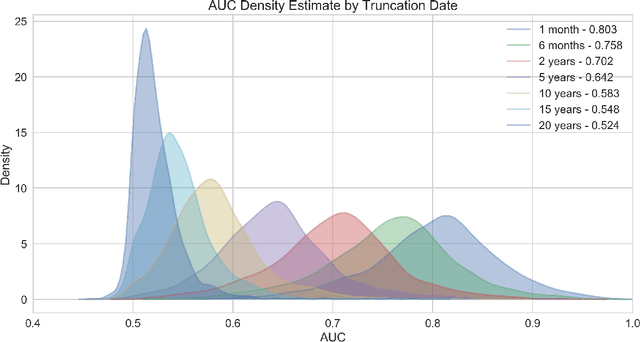
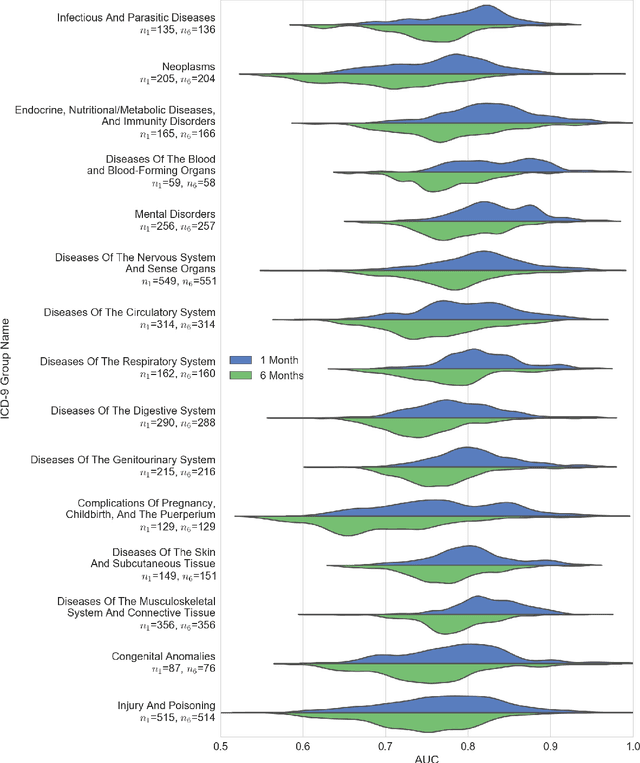

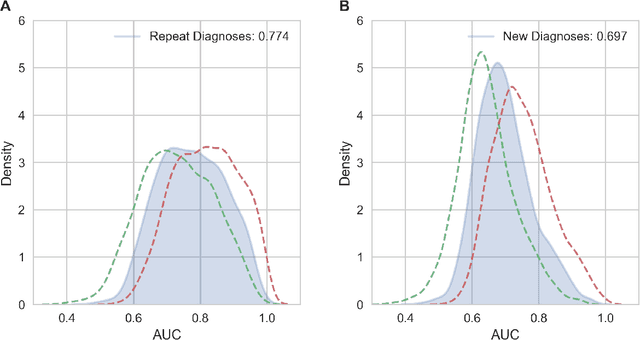
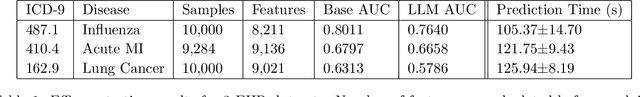
The widespread digitization of patient data via electronic health records (EHRs) has created an unprecedented opportunity to use machine learning algorithms to better predict disease risk at the patient level. Although predictive models have previously been constructed for a few important diseases, such as breast cancer and myocardial infarction, we currently know very little about how accurately the risk for most diseases or events can be predicted, and how far in advance. Machine learning algorithms use training data rather than preprogrammed rules to make predictions and are well suited for the complex task of disease prediction. Although there are thousands of conditions and illnesses patients can encounter, no prior research simultaneously predicts risks for thousands of diagnosis codes and thereby establishes a comprehensive patient risk profile. Here we show that such pandiagnostic prediction is possible with a high level of performance across diagnosis codes. For the tasks of predicting diagnosis risks both 1 and 6 months in advance, we achieve average areas under the receiver operating characteristic curve (AUCs) of 0.803 and 0.758, respectively, across thousands of prediction tasks. Finally, our research contributes a new clinical prediction dataset in which researchers can explore how well a diagnosis can be predicted and what health factors are most useful for prediction. For the first time, we can get a much more complete picture of how well risks for thousands of different diagnosis codes can be predicted.
Machine Learning to Predict Developmental Neurotoxicity with High-throughput Data from 2D Bio-engineered Tissues
May 06, 2019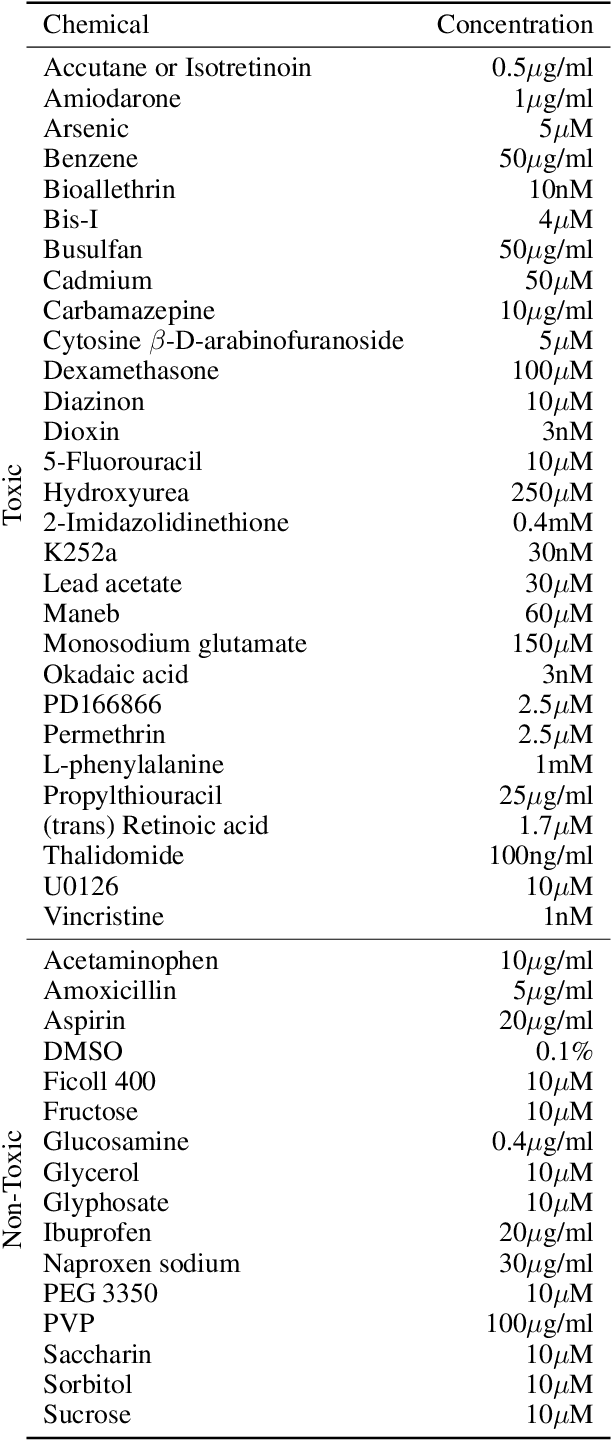
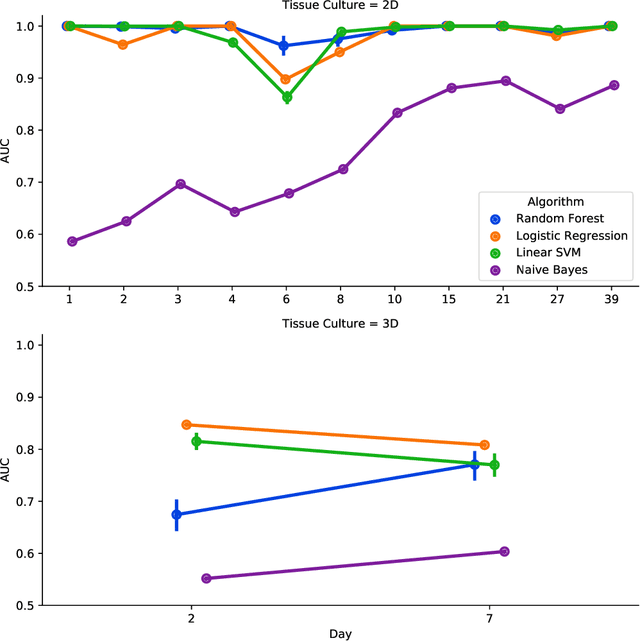
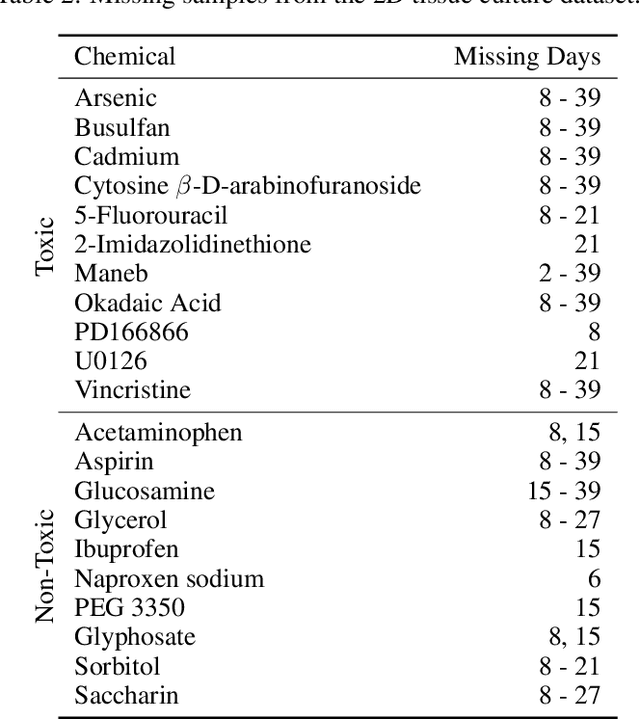
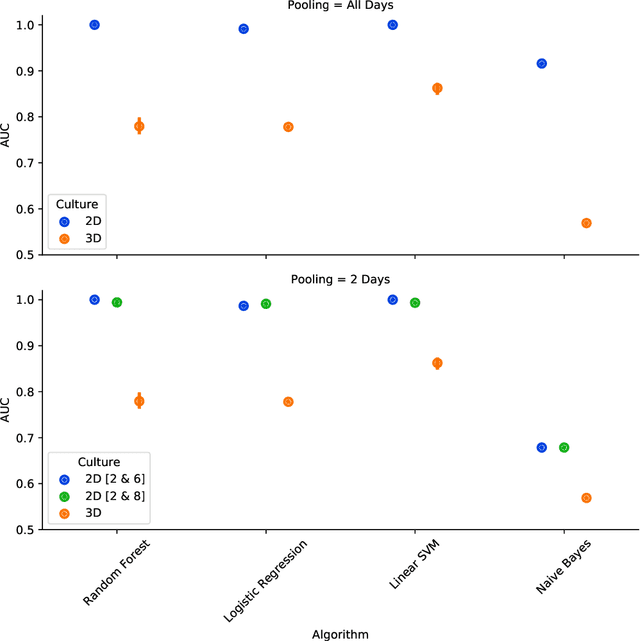
There is a growing need for fast and accurate methods for testing developmental neurotoxicity across several chemical exposure sources. Current approaches, such as in vivo animal studies, and assays of animal and human primary cell cultures, suffer from challenges related to time, cost, and applicability to human physiology. We previously demonstrated success employing machine learning to predict developmental neurotoxicity using gene expression data collected from human 3D tissue models exposed to various compounds. The 3D model is biologically similar to developing neural structures, but its complexity necessitates extensive expertise and effort to employ. By instead focusing solely on constructing an assay of developmental neurotoxicity, we propose that a simpler 2D tissue model may prove sufficient. We thus compare the accuracy of predictive models trained on data from a 2D tissue model with those trained on data from a 3D tissue model, and find the 2D model to be substantially more accurate. Furthermore, we find the 2D model to be more robust under stringent gene set selection, whereas the 3D model suffers substantial accuracy degradation. While both approaches have advantages and disadvantages, we propose that our described 2D approach could be a valuable tool for decision makers when prioritizing neurotoxicity screening.
Privacy-Preserving Collaborative Prediction using Random Forests
Nov 21, 2018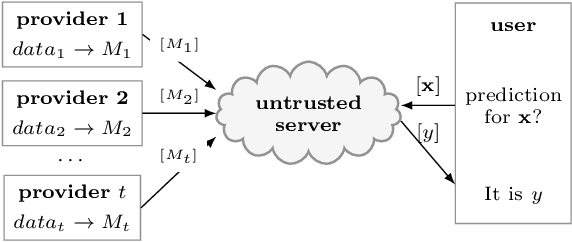
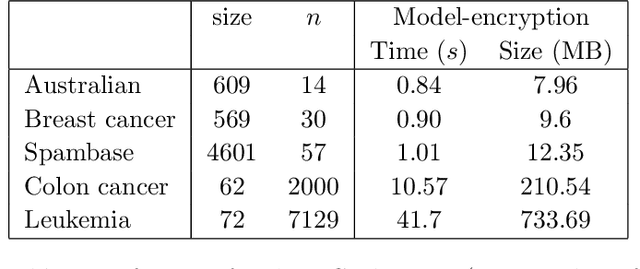
We study the problem of privacy-preserving machine learning (PPML) for ensemble methods, focusing our effort on random forests. In collaborative analysis, PPML attempts to solve the conflict between the need for data sharing and privacy. This is especially important in privacy sensitive applications such as learning predictive models for clinical decision support from EHR data from different clinics, where each clinic has a responsibility for its patients' privacy. We propose a new approach for ensemble methods: each entity learns a model, from its own data, and then when a client asks the prediction for a new private instance, the answers from all the locally trained models are used to compute the prediction in such a way that no extra information is revealed. We implement this approach for random forests and we demonstrate its high efficiency and potential accuracy benefit via experiments on real-world datasets, including actual EHR data.
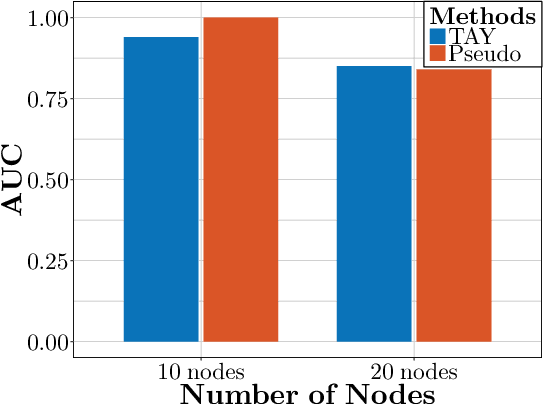
An Efficient Pseudo-likelihood Method for Sparse Binary Pairwise Markov Network Estimation
Apr 07, 2017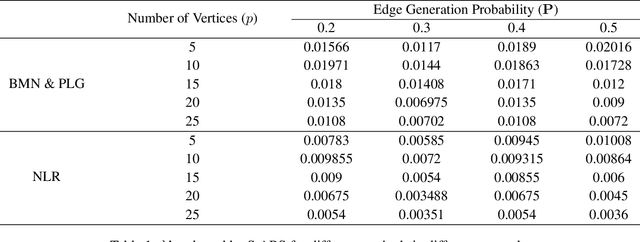
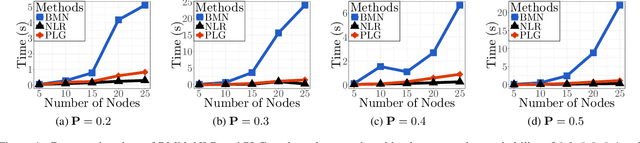
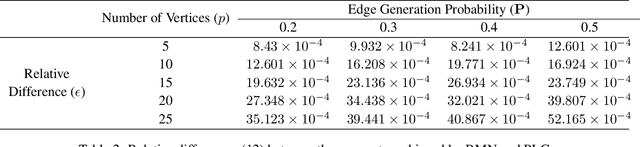
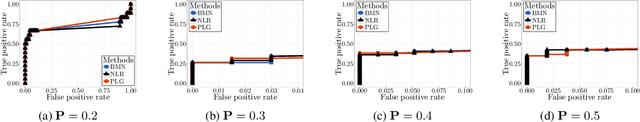
The pseudo-likelihood method is one of the most popular algorithms for learning sparse binary pairwise Markov networks. In this paper, we formulate the $L_1$ regularized pseudo-likelihood problem as a sparse multiple logistic regression problem. In this way, many insights and optimization procedures for sparse logistic regression can be applied to the learning of discrete Markov networks. Specifically, we use the coordinate descent algorithm for generalized linear models with convex penalties, combined with strong screening rules, to solve the pseudo-likelihood problem with $L_1$ regularization. Therefore a substantial speedup without losing any accuracy can be achieved. Furthermore, this method is more stable than the node-wise logistic regression approach on unbalanced high-dimensional data when penalized by small regularization parameters. Thorough numerical experiments on simulated data and real world data demonstrate the advantages of the proposed method.
Computational Drug Repositioning Using Continuous Self-controlled Case Series
Apr 20, 2016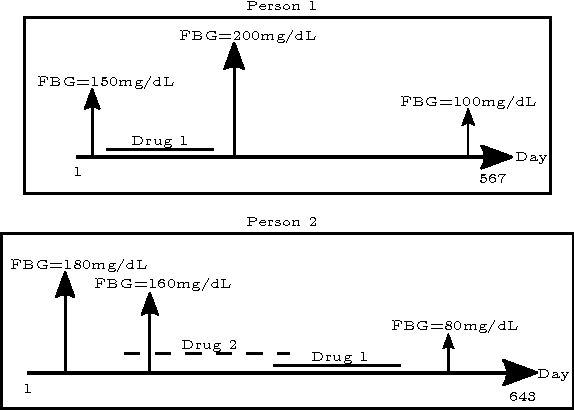
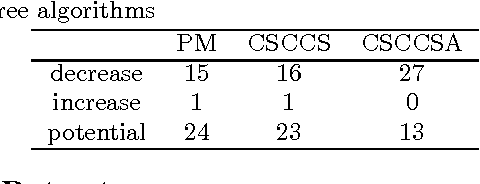
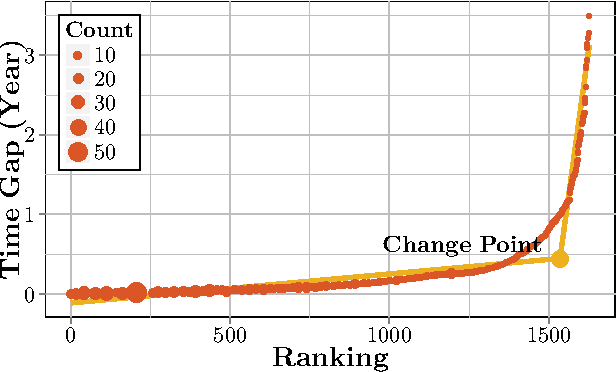
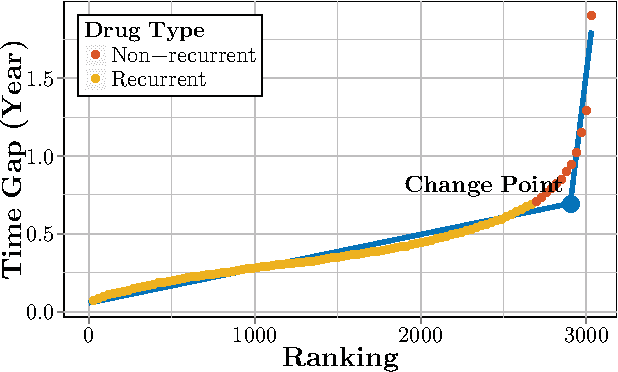
Computational Drug Repositioning (CDR) is the task of discovering potential new indications for existing drugs by mining large-scale heterogeneous drug-related data sources. Leveraging the patient-level temporal ordering information between numeric physiological measurements and various drug prescriptions provided in Electronic Health Records (EHRs), we propose a Continuous Self-controlled Case Series (CSCCS) model for CDR. As an initial evaluation, we look for drugs that can control Fasting Blood Glucose (FBG) level in our experiments. Applying CSCCS to the Marshfield Clinic EHR, well-known drugs that are indicated for controlling blood glucose level are rediscovered. Furthermore, some drugs with recent literature support for the potential effect of blood glucose level control are also identified.
CLP(BN): Constraint Logic Programming for Probabilistic Knowledge
Oct 19, 2012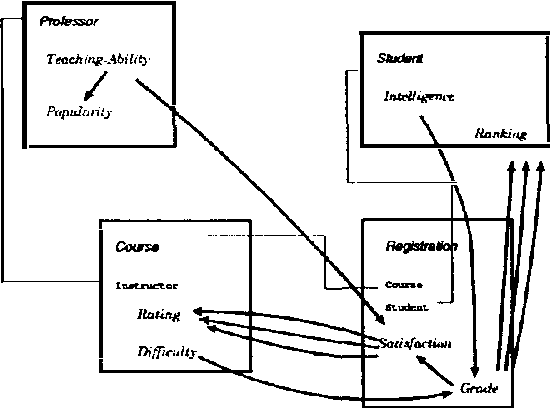
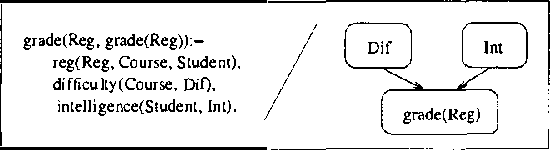
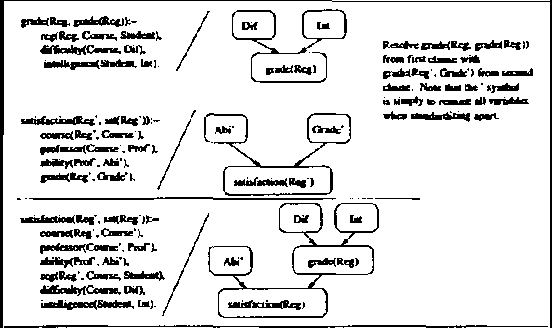
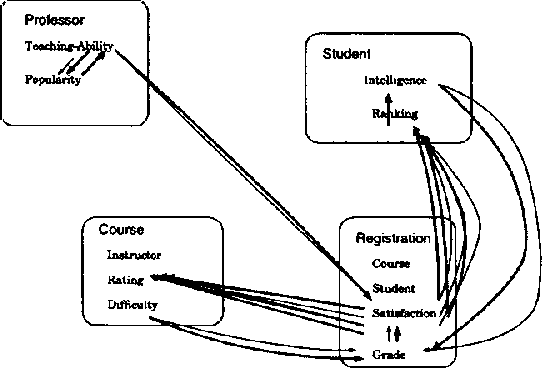
We present CLP(BN), a novel approach that aims at expressing Bayesian networks through the constraint logic programming framework. Arguably, an important limitation of traditional Bayesian networks is that they are propositional, and thus cannot represent relations between multiple similar objects in multiple contexts. Several researchers have thus proposed first-order languages to describe such networks. Namely, one very successful example of this approach are the Probabilistic Relational Models (PRMs), that combine Bayesian networks with relational database technology. The key difficulty that we had to address when designing CLP(cal{BN}) is that logic based representations use ground terms to denote objects. With probabilitic data, we need to be able to uniquely represent an object whose value we are not sure about. We use {sl Skolem functions} as unique new symbols that uniquely represent objects with unknown value. The semantics of CLP(cal{BN}) programs then naturally follow from the general framework of constraint logic programming, as applied to a specific domain where we have probabilistic data. This paper introduces and defines CLP(cal{BN}), and it describes an implementation and initial experiments. The paper also shows how CLP(cal{BN}) relates to Probabilistic Relational Models (PRMs), Ngo and Haddawys Probabilistic Logic Programs, AND Kersting AND De Raedts Bayesian Logic Programs.