 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOpen Source Infrastructure for Differentiable Density Functional Theory
Sep 27, 2023



Learning exchange correlation functionals, used in quantum chemistry calculations, from data has become increasingly important in recent years, but training such a functional requires sophisticated software infrastructure. For this reason, we build open source infrastructure to train neural exchange correlation functionals. We aim to standardize the processing pipeline by adapting state-of-the-art techniques from work done by multiple groups. We have open sourced the model in the DeepChem library to provide a platform for additional research on differentiable quantum chemistry methods.
ChemBERTa-2: Towards Chemical Foundation Models
Sep 05, 2022
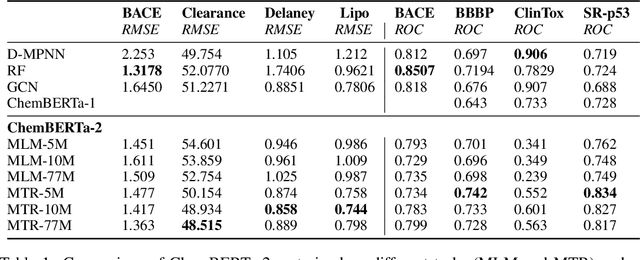
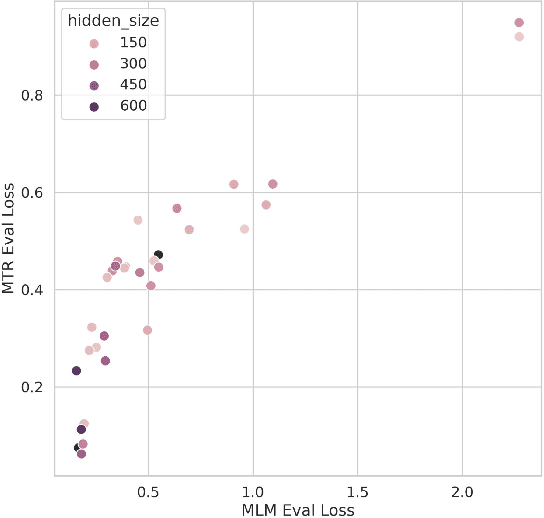
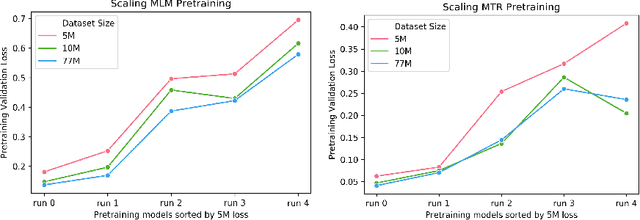
Large pretrained models such as GPT-3 have had tremendous impact on modern natural language processing by leveraging self-supervised learning to learn salient representations that can be used to readily finetune on a wide variety of downstream tasks. We investigate the possibility of transferring such advances to molecular machine learning by building a chemical foundation model, ChemBERTa-2, using the language of SMILES. While labeled data for molecular prediction tasks is typically scarce, libraries of SMILES strings are readily available. In this work, we build upon ChemBERTa by optimizing the pretraining process. We compare multi-task and self-supervised pretraining by varying hyperparameters and pretraining dataset size, up to 77M compounds from PubChem. To our knowledge, the 77M set constitutes one of the largest datasets used for molecular pretraining to date. We find that with these pretraining improvements, we are competitive with existing state-of-the-art architectures on the MoleculeNet benchmark suite. We analyze the degree to which improvements in pretraining translate to improvement on downstream tasks.
Score-Based Generative Models for Molecule Generation
Mar 07, 2022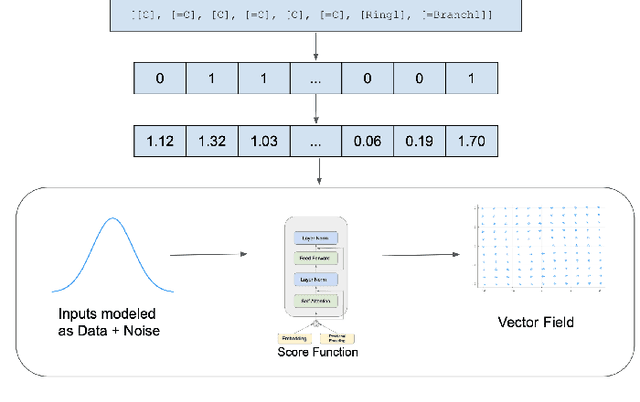
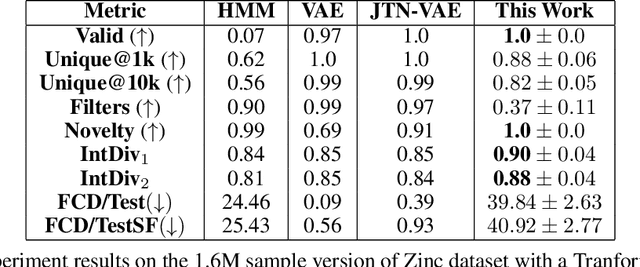
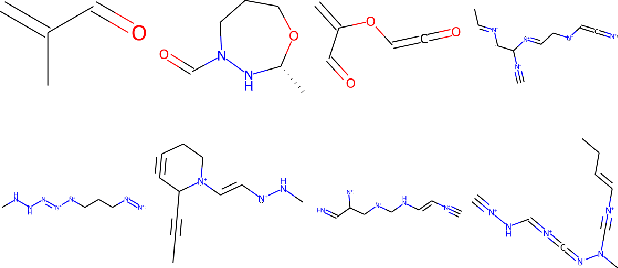
Recent advances in generative models have made exploring design spaces easier for de novo molecule generation. However, popular generative models like GANs and normalizing flows face challenges such as training instabilities due to adversarial training and architectural constraints, respectively. Score-based generative models sidestep these challenges by modelling the gradient of the log probability density using a score function approximation, as opposed to modelling the density function directly, and sampling from it using annealed Langevin Dynamics. We believe that score-based generative models could open up new opportunities in molecule generation due to their architectural flexibility, such as replacing the score function with an SE(3) equivariant model. In this work, we lay the foundations by testing the efficacy of score-based models for molecule generation. We train a Transformer-based score function on Self-Referencing Embedded Strings (SELFIES) representations of 1.5 million samples from the ZINC dataset and use the Moses benchmarking framework to evaluate the generated samples on a suite of metrics.
FastFlows: Flow-Based Models for Molecular Graph Generation
Jan 28, 2022



We propose a framework using normalizing-flow based models, SELF-Referencing Embedded Strings, and multi-objective optimization that efficiently generates small molecules. With an initial training set of only 100 small molecules, FastFlows generates thousands of chemically valid molecules in seconds. Because of the efficient sampling, substructure filters can be applied as desired to eliminate compounds with unreasonable moieties. Using easily computable and learned metrics for druglikeness, synthetic accessibility, and synthetic complexity, we perform a multi-objective optimization to demonstrate how FastFlows functions in a high-throughput virtual screening context. Our model is significantly simpler and easier to train than autoregressive molecular generative models, and enables fast generation and identification of druglike, synthesizable molecules.
Bringing Atomistic Deep Learning to Prime Time
Dec 09, 2021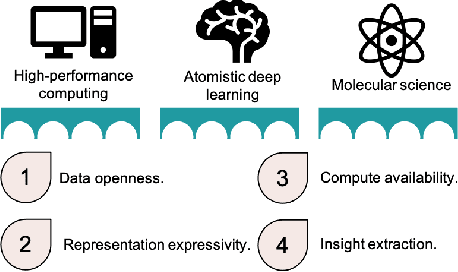
Artificial intelligence has not yet revolutionized the design of materials and molecules. In this perspective, we identify four barriers preventing the integration of atomistic deep learning, molecular science, and high-performance computing. We outline focused research efforts to address the opportunities presented by these challenges.
Differentiable Physics: A Position Piece
Sep 14, 2021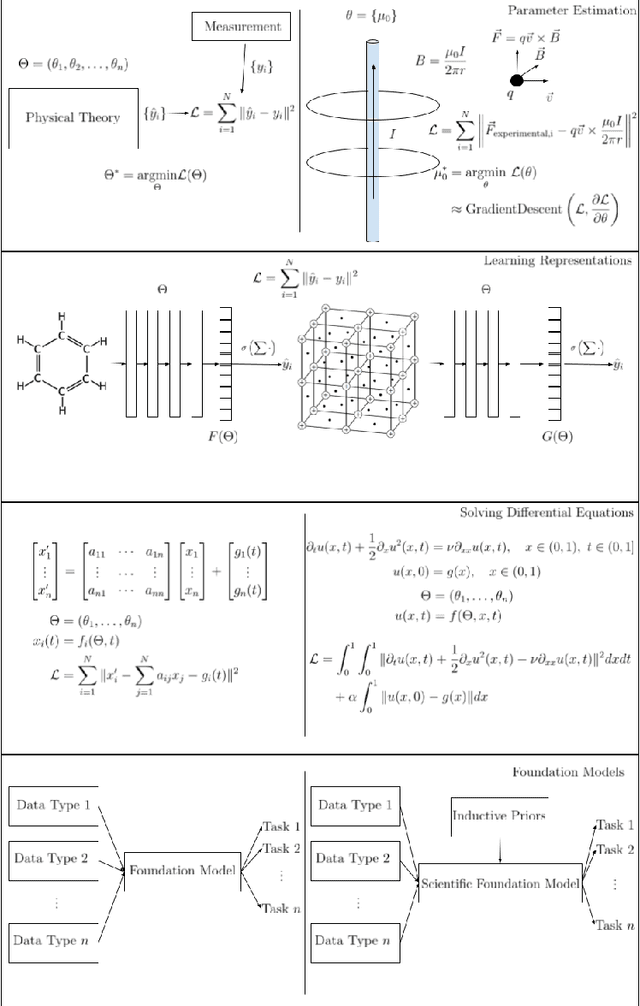
Differentiable physics provides a new approach for modeling and understanding the physical systems by pairing the new technology of differentiable programming with classical numerical methods for physical simulation. We survey the rapidly growing literature of differentiable physics techniques and highlight methods for parameter estimation, learning representations, solving differential equations, and developing what we call scientific foundation models using data and inductive priors. We argue that differentiable physics offers a new paradigm for modeling physical phenomena by combining classical analytic solutions with numerical methodology using the bridge of differentiable programming.
ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction
Oct 23, 2020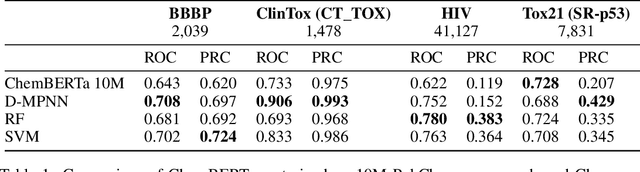
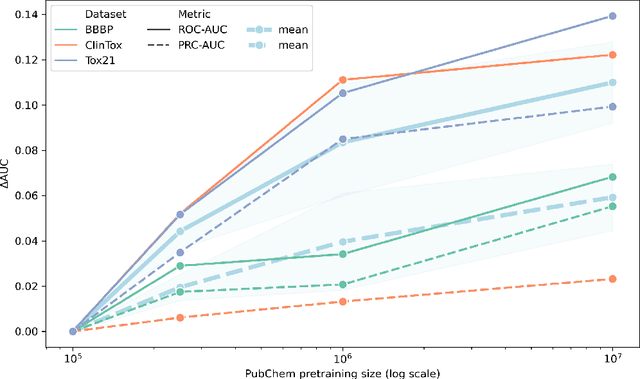
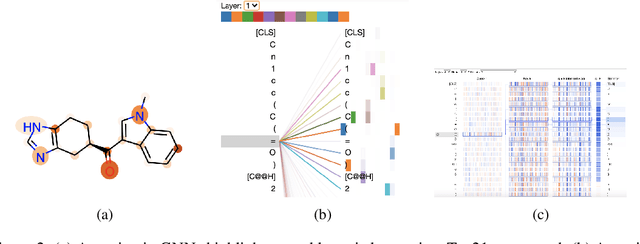
GNNs and chemical fingerprints are the predominant approaches to representing molecules for property prediction. However, in NLP, transformers have become the de-facto standard for representation learning thanks to their strong downstream task transfer. In parallel, the software ecosystem around transformers is maturing rapidly, with libraries like HuggingFace and BertViz enabling streamlined training and introspection. In this work, we make one of the first attempts to systematically evaluate transformers on molecular property prediction tasks via our ChemBERTa model. ChemBERTa scales well with pretraining dataset size, offering competitive downstream performance on MoleculeNet and useful attention-based visualization modalities. Our results suggest that transformers offer a promising avenue of future work for molecular representation learning and property prediction. To facilitate these efforts, we release a curated dataset of 77M SMILES from PubChem suitable for large-scale self-supervised pretraining.
AMPL: A Data-Driven Modeling Pipeline for Drug Discovery
Nov 14, 2019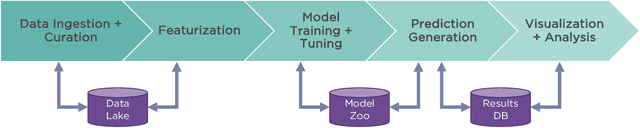
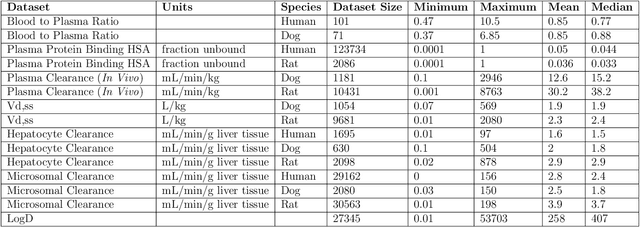
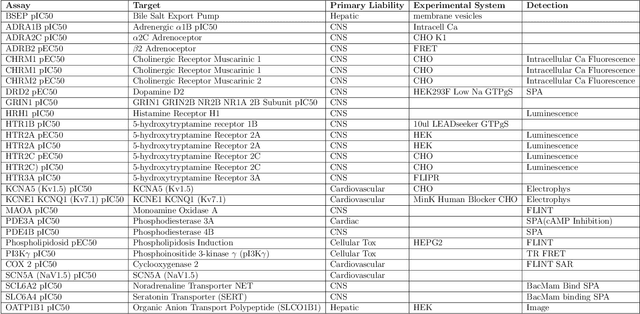
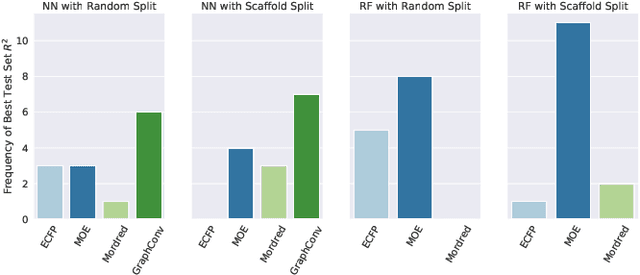
One of the key requirements for incorporating machine learning into the drug discovery process is complete reproducibility and traceability of the model building and evaluation process. With this in mind, we have developed an end-to-end modular and extensible software pipeline for building and sharing machine learning models that predict key pharma-relevant parameters. The ATOM Modeling PipeLine, or AMPL, extends the functionality of the open source library DeepChem and supports an array of machine learning and molecular featurization tools. We have benchmarked AMPL on a large collection of pharmaceutical datasets covering a wide range of parameters. As a result of these comprehensive experiments, we have found that physicochemical descriptors and deep learning-based graph representations significantly outperform traditional fingerprints in the characterization of molecular features. We have also found that dataset size is directly correlated to prediction performance, and that single-task deep learning models only outperform shallow learners if there is sufficient data. Likewise, dataset size has a direct impact on model predictivity, independent of comprehensive hyperparameter model tuning. Our findings point to the need for public dataset integration or multi-task/transfer learning approaches. Lastly, we found that uncertainty quantification (UQ) analysis may help identify model error; however, efficacy of UQ to filter predictions varies considerably between datasets and featurization/model types. AMPL is open source and available for download at http://github.com/ATOMconsortium/AMPL.
MoleculeNet: A Benchmark for Molecular Machine Learning
Oct 26, 2018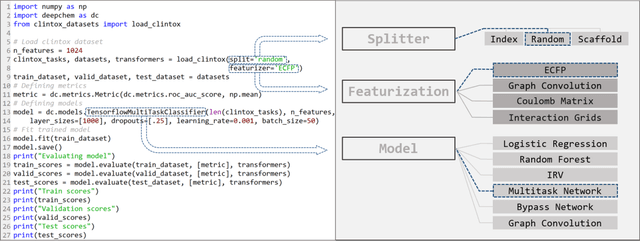
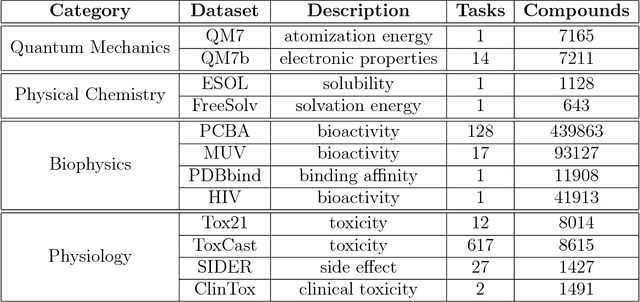
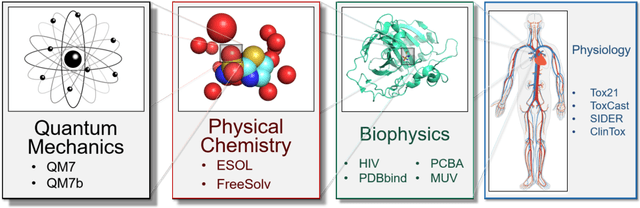
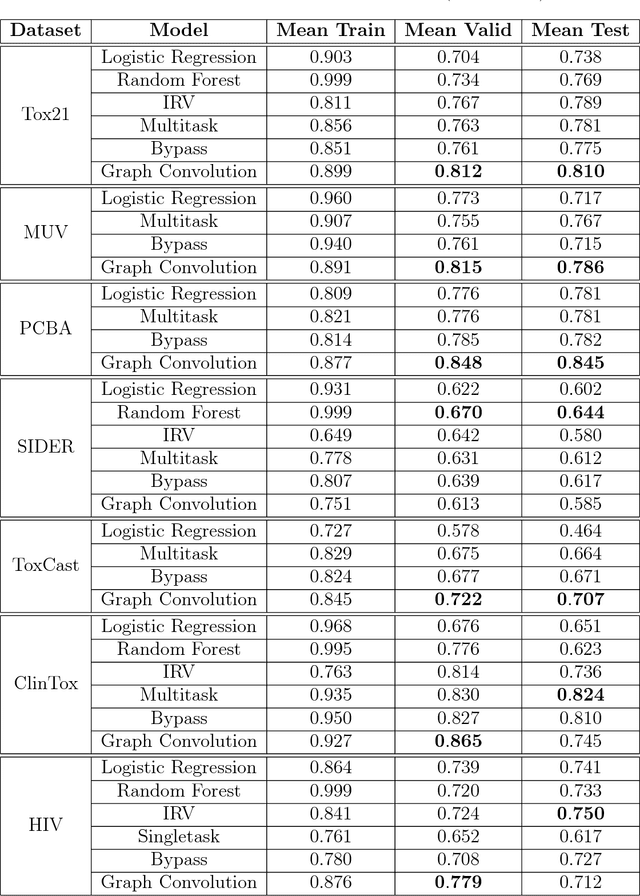
Molecular machine learning has been maturing rapidly over the last few years. Improved methods and the presence of larger datasets have enabled machine learning algorithms to make increasingly accurate predictions about molecular properties. However, algorithmic progress has been limited due to the lack of a standard benchmark to compare the efficacy of proposed methods; most new algorithms are benchmarked on different datasets making it challenging to gauge the quality of proposed methods. This work introduces MoleculeNet, a large scale benchmark for molecular machine learning. MoleculeNet curates multiple public datasets, establishes metrics for evaluation, and offers high quality open-source implementations of multiple previously proposed molecular featurization and learning algorithms (released as part of the DeepChem open source library). MoleculeNet benchmarks demonstrate that learnable representations are powerful tools for molecular machine learning and broadly offer the best performance. However, this result comes with caveats. Learnable representations still struggle to deal with complex tasks under data scarcity and highly imbalanced classification. For quantum mechanical and biophysical datasets, the use of physics-aware featurizations can be more important than choice of particular learning algorithm.
PotentialNet for Molecular Property Prediction
Oct 22, 2018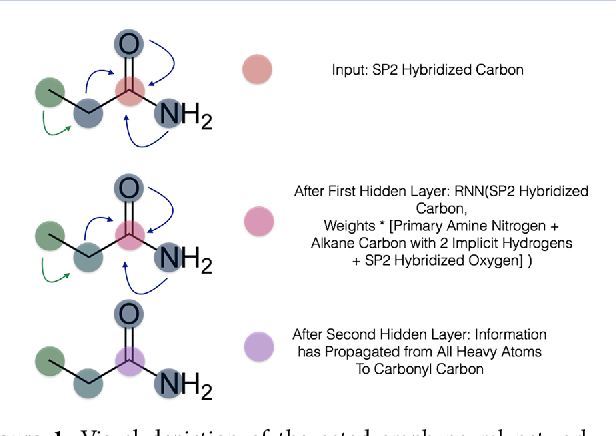
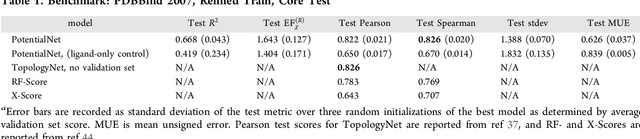
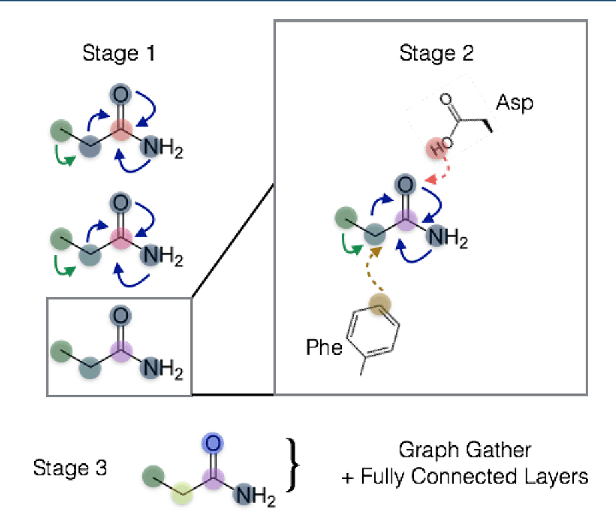
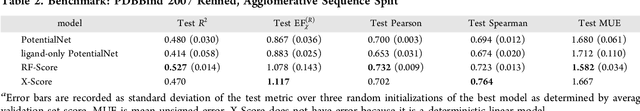
The arc of drug discovery entails a multiparameter optimization problem spanning vast length scales. They key parameters range from solubility (angstroms) to protein-ligand binding (nanometers) to in vivo toxicity (meters). Through feature learning---instead of feature engineering---deep neural networks promise to outperform both traditional physics-based and knowledge-based machine learning models for predicting molecular properties pertinent to drug discovery. To this end, we present the PotentialNet family of graph convolutions. These models are specifically designed for and achieve state-of-the-art performance for protein-ligand binding affinity. We further validate these deep neural networks by setting new standards of performance in several ligand-based tasks. In parallel, we introduce a new metric, the Regression Enrichment Factor $EF_\chi^{(R)}$, to measure the early enrichment of computational models for chemical data. Finally, we introduce a cross-validation strategy based on structural homology clustering that can more accurately measure model generalizability, which crucially distinguishes the aims of machine learning for drug discovery from standard machine learning tasks.