 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChemellia: An Ecosystem for Atomistic Scientific Machine Learning
May 19, 2023


Chemellia is an open-source framework for atomistic machine learning in the Julia programming language. The framework takes advantage of Julia's high speed as well as the ability to share and reuse code and interfaces through the paradigm of multiple dispatch. Chemellia is designed to make use of existing interfaces and avoid ``reinventing the wheel'' wherever possible. A key aspect of the Chemellia ecosystem is the ChemistryFeaturization interface for defining and encoding features -- it is designed to maximize interoperability between featurization schemes and elements thereof, to maintain provenance of encoded features, and to ensure easy decodability and reconfigurability to enable feature engineering experiments. This embodies the overall design principles of the Chemellia ecosystem: separation of concerns, interoperability, and transparency. We illustrate these principles by discussing the implementation of crystal graph convolutional neural networks for material property prediction.
Score-Based Generative Models for Molecule Generation
Mar 07, 2022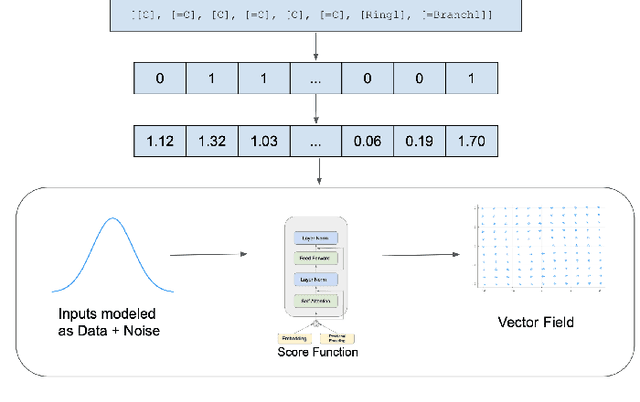
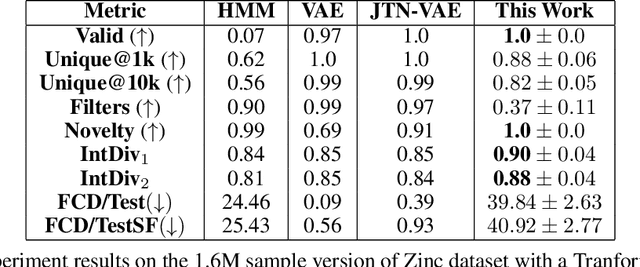
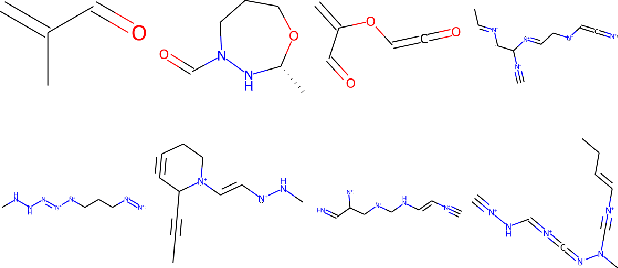
Recent advances in generative models have made exploring design spaces easier for de novo molecule generation. However, popular generative models like GANs and normalizing flows face challenges such as training instabilities due to adversarial training and architectural constraints, respectively. Score-based generative models sidestep these challenges by modelling the gradient of the log probability density using a score function approximation, as opposed to modelling the density function directly, and sampling from it using annealed Langevin Dynamics. We believe that score-based generative models could open up new opportunities in molecule generation due to their architectural flexibility, such as replacing the score function with an SE(3) equivariant model. In this work, we lay the foundations by testing the efficacy of score-based models for molecule generation. We train a Transformer-based score function on Self-Referencing Embedded Strings (SELFIES) representations of 1.5 million samples from the ZINC dataset and use the Moses benchmarking framework to evaluate the generated samples on a suite of metrics.
ACED: Accelerated Computational Electrochemical systems Discovery
Nov 10, 2020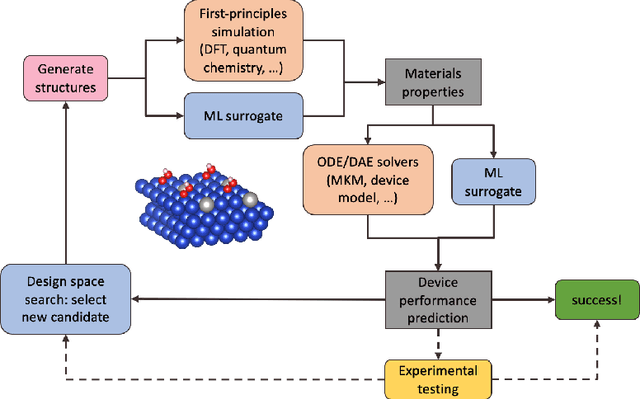
Large-scale electrification is vital to addressing the climate crisis, but many engineering challenges remain to fully electrifying both the chemical industry and transportation. In both of these areas, new electrochemical materials and systems will be critical, but developing these systems currently relies heavily on computationally expensive first-principles simulations as well as human-time-intensive experimental trial and error. We propose to develop an automated workflow that accelerates these computational steps by introducing both automated error handling in generating the first-principles training data as well as physics-informed machine learning surrogates to further reduce computational cost. It will also have the capacity to include automated experiments "in the loop" in order to dramatically accelerate the overall materials discovery pipeline.
Fashionable Modelling with Flux
Nov 01, 2018
Machine learning as a discipline has seen an incredible surge of interest in recent years due in large part to a perfect storm of new theory, superior tooling, renewed interest in its capabilities. We present in this paper a framework named Flux that shows how further refinement of the core ideas of machine learning, built upon the foundation of the Julia programming language, can yield an environment that is simple, easily modifiable, and performant. We detail the fundamental principles of Flux as a framework for differentiable programming, give examples of models that are implemented within Flux to display many of the language and framework-level features that contribute to its ease of use and high productivity, display internal compiler techniques used to enable the acceleration and performance that lies at the heart of Flux, and finally give an overview of the larger ecosystem that Flux fits inside of.