 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMotif-based Graph Self-Supervised Learning for Molecular Property Prediction
Paper and Code
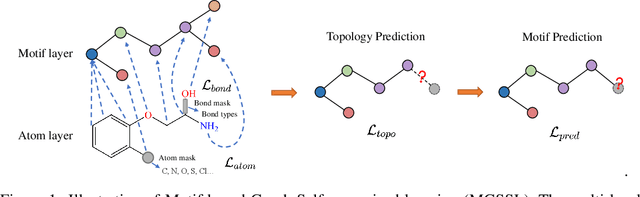
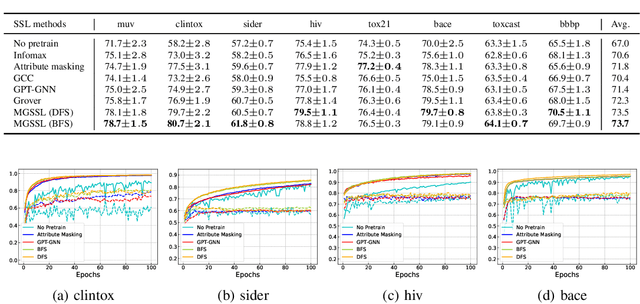
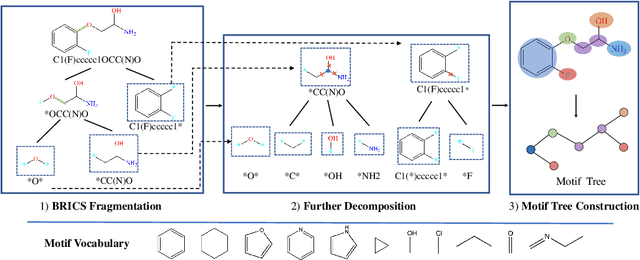
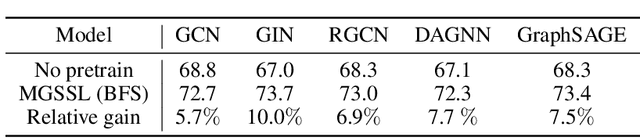
Predicting molecular properties with data-driven methods has drawn much attention in recent years. Particularly, Graph Neural Networks (GNNs) have demonstrated remarkable success in various molecular generation and prediction tasks. In cases where labeled data is scarce, GNNs can be pre-trained on unlabeled molecular data to first learn the general semantic and structural information before being fine-tuned for specific tasks. However, most existing self-supervised pre-training frameworks for GNNs only focus on node-level or graph-level tasks. These approaches cannot capture the rich information in subgraphs or graph motifs. For example, functional groups (frequently-occurred subgraphs in molecular graphs) often carry indicative information about the molecular properties. To bridge this gap, we propose Motif-based Graph Self-supervised Learning (MGSSL) by introducing a novel self-supervised motif generation framework for GNNs. First, for motif extraction from molecular graphs, we design a molecule fragmentation method that leverages a retrosynthesis-based algorithm BRICS and additional rules for controlling the size of motif vocabulary. Second, we design a general motif-based generative pre-training framework in which GNNs are asked to make topological and label predictions. This generative framework can be implemented in two different ways, i.e., breadth-first or depth-first. Finally, to take the multi-scale information in molecular graphs into consideration, we introduce a multi-level self-supervised pre-training. Extensive experiments on various downstream benchmark tasks show that our methods outperform all state-of-the-art baselines.