 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDistance-aware Molecule Graph Attention Network for Drug-Target Binding Affinity Prediction
Paper and Code
Dec 17, 2020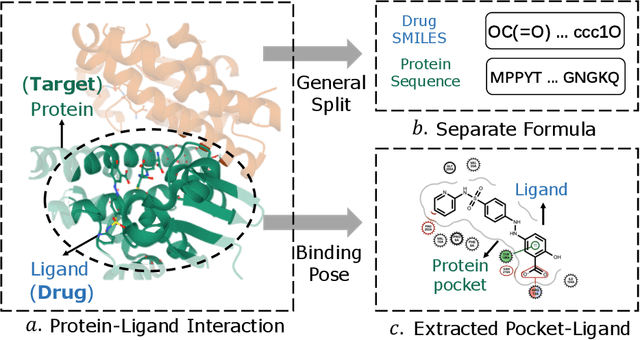

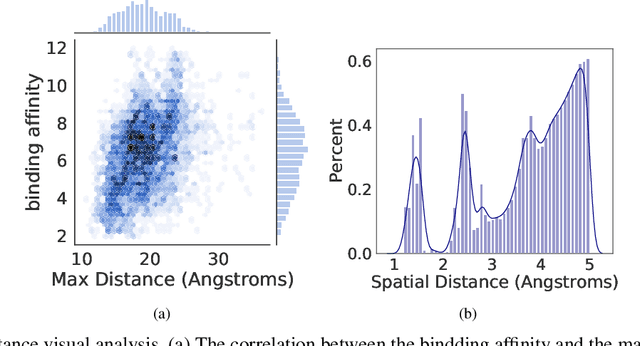
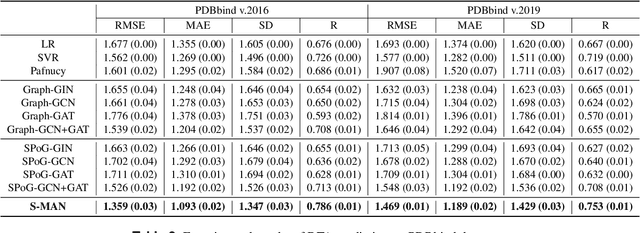
Accurately predicting the binding affinity between drugs and proteins is an essential step for computational drug discovery. Since graph neural networks (GNNs) have demonstrated remarkable success in various graph-related tasks, GNNs have been considered as a promising tool to improve the binding affinity prediction in recent years. However, most of the existing GNN architectures can only encode the topological graph structure of drugs and proteins without considering the relative spatial information among their atoms. Whereas, different from other graph datasets such as social networks and commonsense knowledge graphs, the relative spatial position and chemical bonds among atoms have significant impacts on the binding affinity. To this end, in this paper, we propose a diStance-aware Molecule graph Attention Network (S-MAN) tailored to drug-target binding affinity prediction. As a dedicated solution, we first propose a position encoding mechanism to integrate the topological structure and spatial position information into the constructed pocket-ligand graph. Moreover, we propose a novel edge-node hierarchical attentive aggregation structure which has edge-level aggregation and node-level aggregation. The hierarchical attentive aggregation can capture spatial dependencies among atoms, as well as fuse the position-enhanced information with the capability of discriminating multiple spatial relations among atoms. Finally, we conduct extensive experiments on two standard datasets to demonstrate the effectiveness of S-MAN.