 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRetroXpert: Decompose Retrosynthesis Prediction like a Chemist
Paper and Code
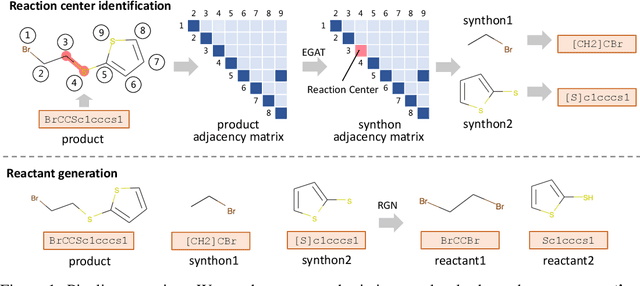
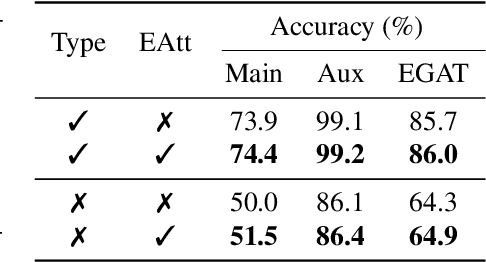
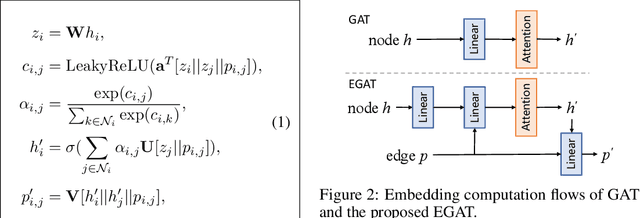
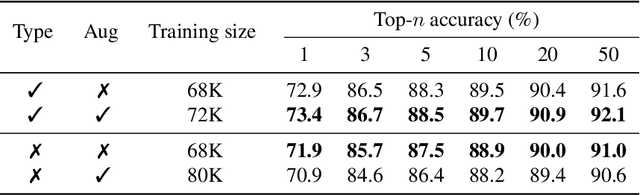
Retrosynthesis is the process of recursively decomposing target molecules into available building blocks. It plays an important role in solving problems in organic synthesis planning. To automate or assist in the retrosynthesis analysis, various retrosynthesis prediction algorithms have been proposed. However, most of them are cumbersome and lack interpretability about their predictions. In this paper, we devise a novel template-free algorithm for automatic retrosynthetic expansion inspired by how chemists approach retrosynthesis prediction. Our method disassembles retrosynthesis into two steps: i) identify the potential reaction center of the target molecule through a novel graph neural network and generate intermediate synthons, and ii) generate the reactants associated with synthons via a robust reactant generation model. While outperforming the state-of-the-art baselines by a significant margin, our model also provides chemically reasonable interpretation.