 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGROVER: Self-supervised Message Passing Transformer on Large-scale Molecular Data
Paper and Code
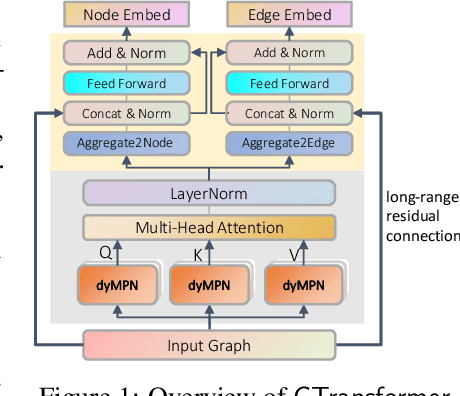
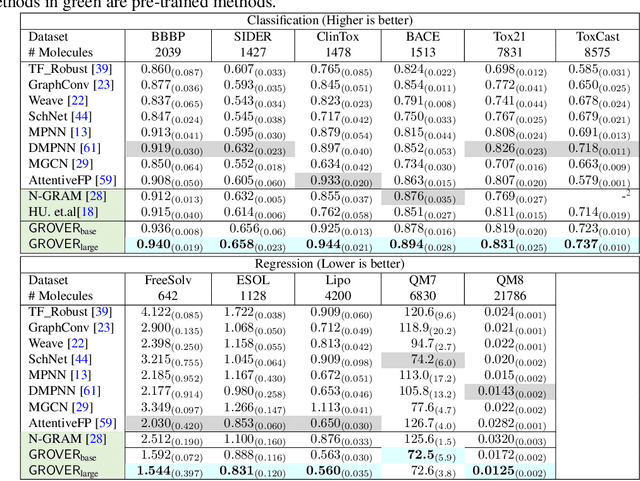
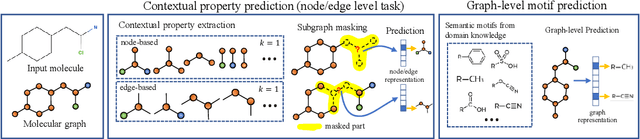
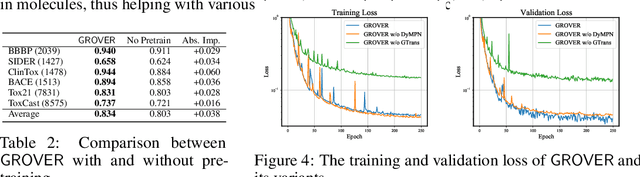
How to obtain informative representations of molecules is a crucial prerequisite in AI-driven drug design and discovery. Recent researches abstract molecules as graphs and employ Graph Neural Networks (GNNs) for task-specific and data-driven molecular representation learning. Nevertheless, two "dark clouds" impede the usage of GNNs in real scenarios: (1) insufficient labeled molecules for supervised training; (2) poor generalization capabilities to new-synthesized molecules. To address them both, we propose a novel molecular representation framework, GROVER, which stands for Graph Representation frOm self-superVised mEssage passing tRansformer. With carefully designed self-supervised tasks in node, edge and graph-level, GROVER can learn rich structural and semantic information of molecules from enormous unlabelled molecular data. Rather, to encode such complex information, GROVER integrates Message Passing Networks with the Transformer-style architecture to deliver a class of more expressive encoders of molecules. The flexibility of GROVER allows it to be trained efficiently on large-scale molecular dataset without requiring any supervision, thus being immunized to the two issues mentioned above. We pre-train GROVER with 100 million parameters on 10 million unlabelled molecules---the biggest GNN and the largest training dataset that we have ever met. We then leverage the pre-trained GROVER to downstream molecular property prediction tasks followed by task-specific fine-tuning, where we observe a huge improvement (more than 6% on average) over current state-of-the-art methods on 11 challenging benchmarks. The insights we gained are that well-designed self-supervision losses and largely-expressive pre-trained models enjoy the significant potential on performance boosting.