 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOpen Materials Generation with Stochastic Interpolants
Feb 04, 2025The discovery of new materials is essential for enabling technological advancements. Computational approaches for predicting novel materials must effectively learn the manifold of stable crystal structures within an infinite design space. We introduce Open Materials Generation (OMG), a unifying framework for the generative design and discovery of inorganic crystalline materials. OMG employs stochastic interpolants (SI) to bridge an arbitrary base distribution to the target distribution of inorganic crystals via a broad class of tunable stochastic processes, encompassing both diffusion models and flow matching as special cases. In this work, we adapt the SI framework by integrating an equivariant graph representation of crystal structures and extending it to account for periodic boundary conditions in unit cell representations. Additionally, we couple the SI flow over spatial coordinates and lattice vectors with discrete flow matching for atomic species. We benchmark OMG's performance on two tasks: Crystal Structure Prediction (CSP) for specified compositions, and 'de novo' generation (DNG) aimed at discovering stable, novel, and unique structures. In our ground-up implementation of OMG, we refine and extend both CSP and DNG metrics compared to previous works. OMG establishes a new state-of-the-art in generative modeling for materials discovery, outperforming purely flow-based and diffusion-based implementations. These results underscore the importance of designing flexible deep learning frameworks to accelerate progress in materials science.
Fine-Tuning Language Models on Multiple Datasets for Citation Intention Classification
Oct 17, 2024



Citation intention Classification (CIC) tools classify citations by their intention (e.g., background, motivation) and assist readers in evaluating the contribution of scientific literature. Prior research has shown that pretrained language models (PLMs) such as SciBERT can achieve state-of-the-art performance on CIC benchmarks. PLMs are trained via self-supervision tasks on a large corpus of general text and can quickly adapt to CIC tasks via moderate fine-tuning on the corresponding dataset. Despite their advantages, PLMs can easily overfit small datasets during fine-tuning. In this paper, we propose a multi-task learning (MTL) framework that jointly fine-tunes PLMs on a dataset of primary interest together with multiple auxiliary CIC datasets to take advantage of additional supervision signals. We develop a data-driven task relation learning (TRL) method that controls the contribution of auxiliary datasets to avoid negative transfer and expensive hyper-parameter tuning. We conduct experiments on three CIC datasets and show that fine-tuning with additional datasets can improve the PLMs' generalization performance on the primary dataset. PLMs fine-tuned with our proposed framework outperform the current state-of-the-art models by 7% to 11% on small datasets while aligning with the best-performing model on a large dataset.
Injecting Domain Knowledge from Empirical Interatomic Potentials to Neural Networks for Predicting Material Properties
Oct 14, 2022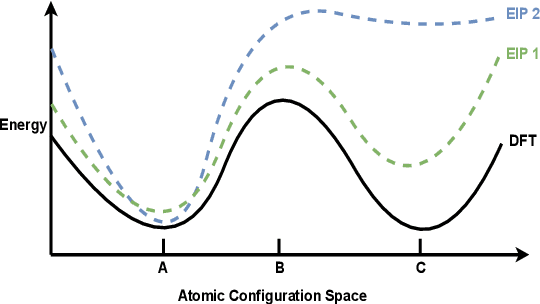
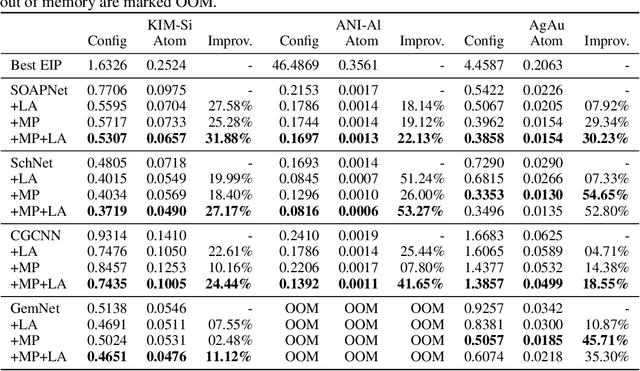
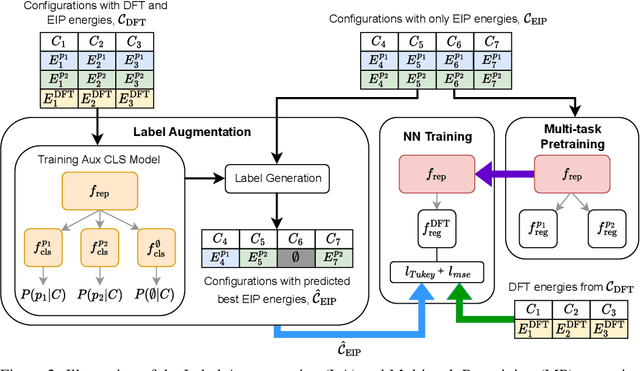
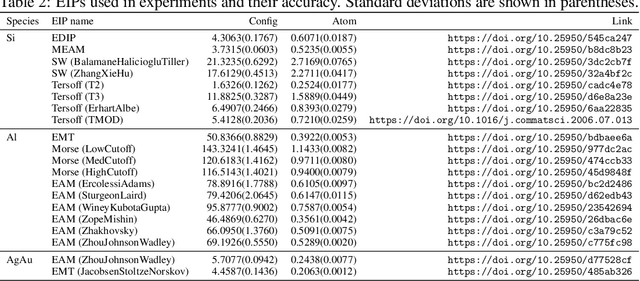
For decades, atomistic modeling has played a crucial role in predicting the behavior of materials in numerous fields ranging from nanotechnology to drug discovery. The most accurate methods in this domain are rooted in first-principles quantum mechanical calculations such as density functional theory (DFT). Because these methods have remained computationally prohibitive, practitioners have traditionally focused on defining physically motivated closed-form expressions known as empirical interatomic potentials (EIPs) that approximately model the interactions between atoms in materials. In recent years, neural network (NN)-based potentials trained on quantum mechanical (DFT-labeled) data have emerged as a more accurate alternative to conventional EIPs. However, the generalizability of these models relies heavily on the amount of labeled training data, which is often still insufficient to generate models suitable for general-purpose applications. In this paper, we propose two generic strategies that take advantage of unlabeled training instances to inject domain knowledge from conventional EIPs to NNs in order to increase their generalizability. The first strategy, based on weakly supervised learning, trains an auxiliary classifier on EIPs and selects the best-performing EIP to generate energies to supplement the ground-truth DFT energies in training the NN. The second strategy, based on transfer learning, first pretrains the NN on a large set of easily obtainable EIP energies, and then fine-tunes it on ground-truth DFT energies. Experimental results on three benchmark datasets demonstrate that the first strategy improves baseline NN performance by 5% to 51% while the second improves baseline performance by up to 55%. Combining them further boosts performance.
Heterogeneous Molecular Graph Neural Networks for Predicting Molecule Properties
Sep 26, 2020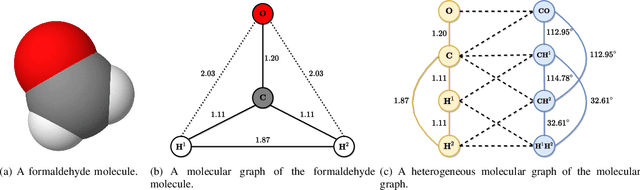
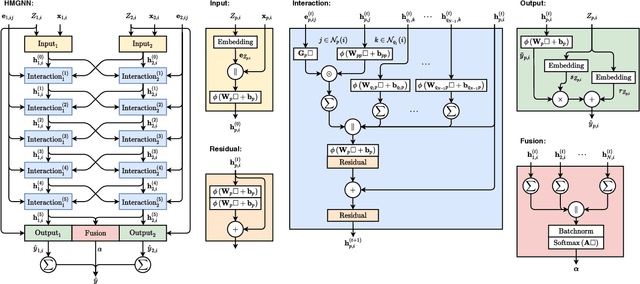
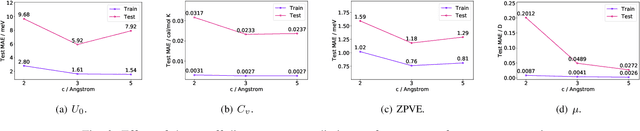
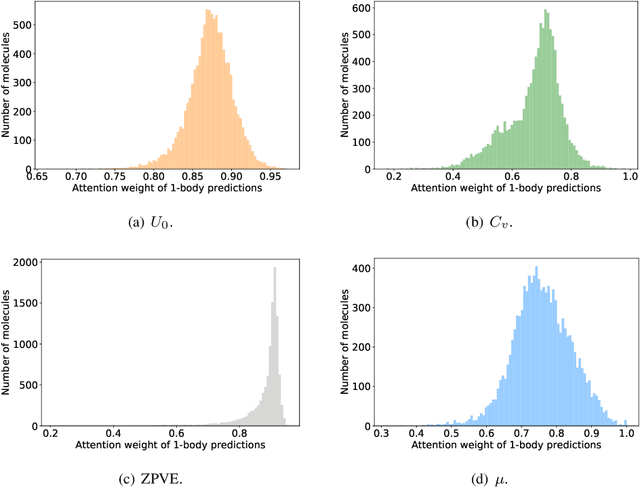
As they carry great potential for modeling complex interactions, graph neural network (GNN)-based methods have been widely used to predict quantum mechanical properties of molecules. Most of the existing methods treat molecules as molecular graphs in which atoms are modeled as nodes. They characterize each atom's chemical environment by modeling its pairwise interactions with other atoms in the molecule. Although these methods achieve a great success, limited amount of works explicitly take many-body interactions, i.e., interactions between three and more atoms, into consideration. In this paper, we introduce a novel graph representation of molecules, heterogeneous molecular graph (HMG) in which nodes and edges are of various types, to model many-body interactions. HMGs have the potential to carry complex geometric information. To leverage the rich information stored in HMGs for chemical prediction problems, we build heterogeneous molecular graph neural networks (HMGNN) on the basis of a neural message passing scheme. HMGNN incorporates global molecule representations and an attention mechanism into the prediction process. The predictions of HMGNN are invariant to translation and rotation of atom coordinates, and permutation of atom indices. Our model achieves state-of-the-art performance in 9 out of 12 tasks on the QM9 dataset.