 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMixture of Rationale: Multi-Modal Reasoning Mixture for Visual Question Answering
Jun 03, 2024Zero-shot visual question answering (VQA) is a challenging task that requires reasoning across modalities. While some existing methods rely on a single rationale within the Chain of Thoughts (CoT) framework, they may fall short of capturing the complexity of the VQA problem. On the other hand, some other methods that use multiple rationales may still suffer from low diversity, poor modality alignment, and inefficient retrieval and fusion. In response to these challenges, we propose \emph{Mixture of Rationales (MoR)}, a novel multi-modal reasoning method that mixes multiple rationales for VQA. MoR uses a single frozen Vision-and-Language Pre-trained Models (VLPM) model to {dynamically generate, retrieve and fuse multi-modal thoughts}. We evaluate MoR on two challenging VQA datasets, i.e. NLVR2 and OKVQA, with two representative backbones OFA and VL-T5. MoR achieves a 12.43\% accuracy improvement on NLVR2, and a 2.45\% accuracy improvement on OKVQA-S( the science and technology category of OKVQA).
Robust Bayesian optimization with reinforcement learned acquisition functions
Oct 02, 2022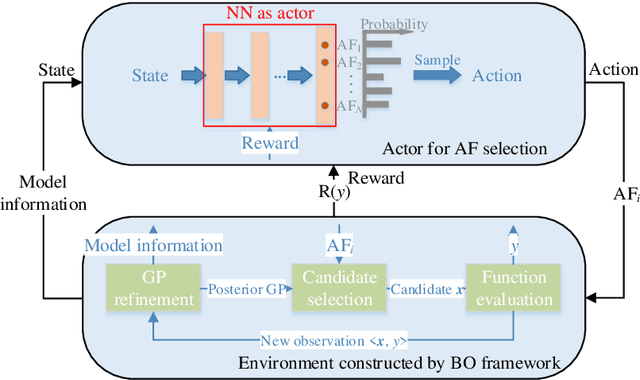
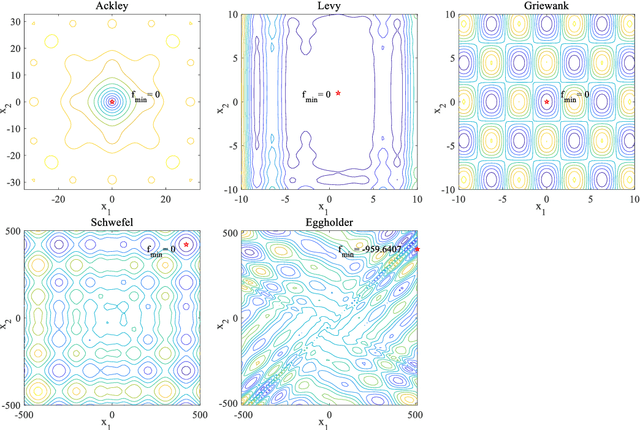
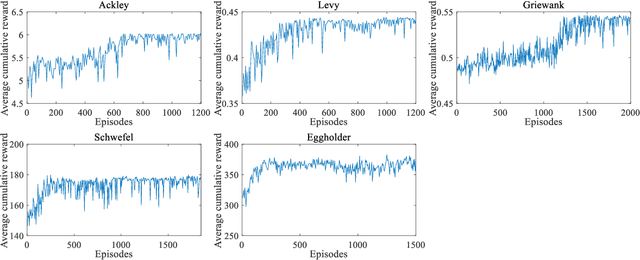
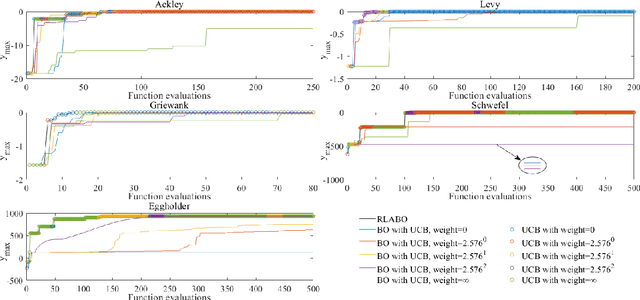
In Bayesian optimization (BO) for expensive black-box optimization tasks, acquisition function (AF) guides sequential sampling and plays a pivotal role for efficient convergence to better optima. Prevailing AFs usually rely on artificial experiences in terms of preferences for exploration or exploitation, which runs a risk of a computational waste or traps in local optima and resultant re-optimization. To address the crux, the idea of data-driven AF selection is proposed, and the sequential AF selection task is further formalized as a Markov decision process (MDP) and resort to powerful reinforcement learning (RL) technologies. Appropriate selection policy for AFs is learned from superior BO trajectories to balance between exploration and exploitation in real time, which is called reinforcement-learning-assisted Bayesian optimization (RLABO). Competitive and robust BO evaluations on five benchmark problems demonstrate RL's recognition of the implicit AF selection pattern and imply the proposal's potential practicality for intelligent AF selection as well as efficient optimization in expensive black-box problems.
Contrastive learning-based computational histopathology predict differential expression of cancer driver genes
Apr 27, 2022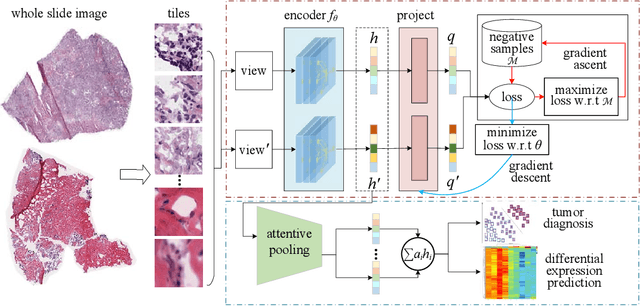
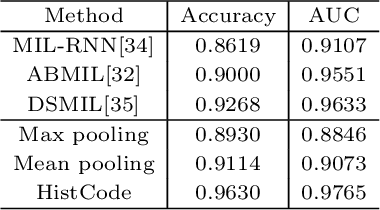
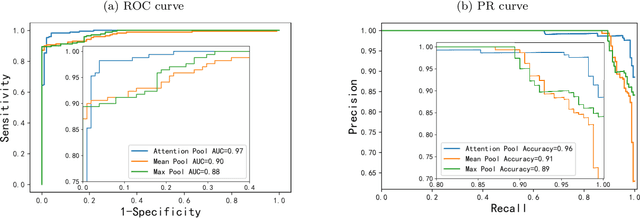
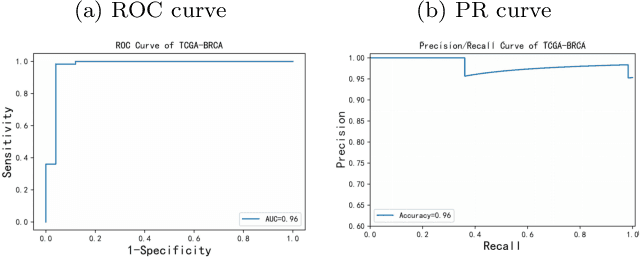
Digital pathological analysis is run as the main examination used for cancer diagnosis. Recently, deep learning-driven feature extraction from pathology images is able to detect genetic variations and tumor environment, but few studies focus on differential gene expression in tumor cells. In this paper, we propose a self-supervised contrastive learning framework, HistCode, to infer differential gene expressions from whole slide images (WSIs). We leveraged contrastive learning on large-scale unannotated WSIs to derive slide-level histopathological feature in latent space, and then transfer it to tumor diagnosis and prediction of differentially expressed cancer driver genes. Our extensive experiments showed that our method outperformed other state-of-the-art models in tumor diagnosis tasks, and also effectively predicted differential gene expressions. Interestingly, we found the higher fold-changed genes can be more precisely predicted. To intuitively illustrate the ability to extract informative features from pathological images, we spatially visualized the WSIs colored by the attentive scores of image tiles. We found that the tumor and necrosis areas were highly consistent with the annotations of experienced pathologists. Moreover, the spatial heatmap generated by lymphocyte-specific gene expression patterns was also consistent with the manually labeled WSI.
Graph2MDA: a multi-modal variational graph embedding model for predicting microbe-drug associations
Aug 14, 2021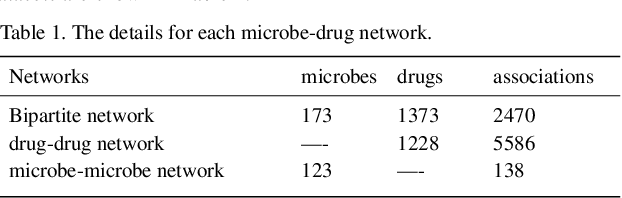

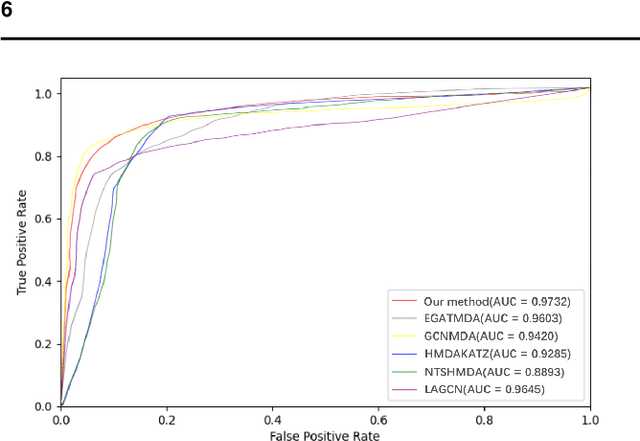

Accumulated clinical studies show that microbes living in humans interact closely with human hosts, and get involved in modulating drug efficacy and drug toxicity. Microbes have become novel targets for the development of antibacterial agents. Therefore, screening of microbe-drug associations can benefit greatly drug research and development. With the increase of microbial genomic and pharmacological datasets, we are greatly motivated to develop an effective computational method to identify new microbe-drug associations. In this paper, we proposed a novel method, Graph2MDA, to predict microbe-drug associations by using variational graph autoencoder (VGAE). We constructed multi-modal attributed graphs based on multiple features of microbes and drugs, such as molecular structures, microbe genetic sequences, and function annotations. Taking as input the multi-modal attribute graphs, VGAE was trained to learn the informative and interpretable latent representations of each node and the whole graph, and then a deep neural network classifier was used to predict microbe-drug associations. The hyperparameter analysis and model ablation studies showed the sensitivity and robustness of our model. We evaluated our method on three independent datasets and the experimental results showed that our proposed method outperformed six existing state-of-the-art methods. We also explored the meaningness of the learned latent representations of drugs and found that the drugs show obvious clustering patterns that are significantly consistent with drug ATC classification. Moreover, we conducted case studies on two microbes and two drugs and found 75\%-95\% predicted associations have been reported in PubMed literature. Our extensive performance evaluations validated the effectiveness of our proposed method.\