 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBohrium + SciMaster: Building the Infrastructure and Ecosystem for Agentic Science at Scale
Dec 23, 2025



AI agents are emerging as a practical way to run multi-step scientific workflows that interleave reasoning with tool use and verification, pointing to a shift from isolated AI-assisted steps toward \emph{agentic science at scale}. This shift is increasingly feasible, as scientific tools and models can be invoked through stable interfaces and verified with recorded execution traces, and increasingly necessary, as AI accelerates scientific output and stresses the peer-review and publication pipeline, raising the bar for traceability and credible evaluation. However, scaling agentic science remains difficult: workflows are hard to observe and reproduce; many tools and laboratory systems are not agent-ready; execution is hard to trace and govern; and prototype AI Scientist systems are often bespoke, limiting reuse and systematic improvement from real workflow signals. We argue that scaling agentic science requires an infrastructure-and-ecosystem approach, instantiated in Bohrium+SciMaster. Bohrium acts as a managed, traceable hub for AI4S assets -- akin to a HuggingFace of AI for Science -- that turns diverse scientific data, software, compute, and laboratory systems into agent-ready capabilities. SciMaster orchestrates these capabilities into long-horizon scientific workflows, on which scientific agents can be composed and executed. Between infrastructure and orchestration, a \emph{scientific intelligence substrate} organizes reusable models, knowledge, and components into executable building blocks for workflow reasoning and action, enabling composition, auditability, and improvement through use. We demonstrate this stack with eleven representative master agents in real workflows, achieving orders-of-magnitude reductions in end-to-end scientific cycle time and generating execution-grounded signals from real workloads at multi-million scale.
Inverse Knowledge Search over Verifiable Reasoning: Synthesizing a Scientific Encyclopedia from a Long Chains-of-Thought Knowledge Base
Oct 30, 2025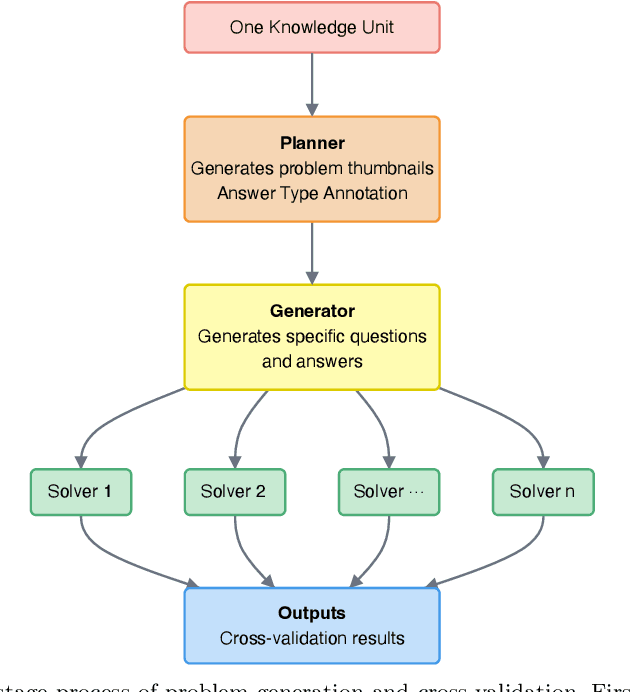
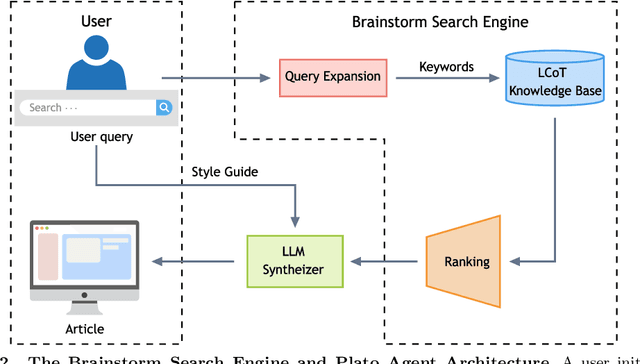
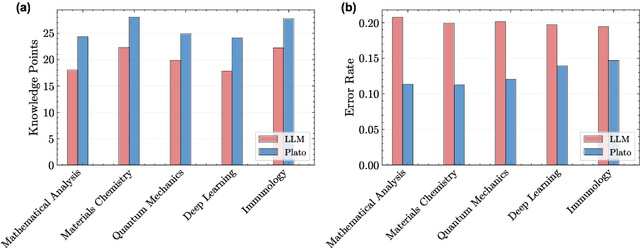
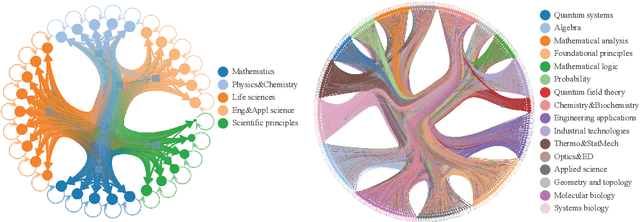
Most scientific materials compress reasoning, presenting conclusions while omitting the derivational chains that justify them. This compression hinders verification by lacking explicit, step-wise justifications and inhibits cross-domain links by collapsing the very pathways that establish the logical and causal connections between concepts. We introduce a scalable framework that decompresses scientific reasoning, constructing a verifiable Long Chain-of-Thought (LCoT) knowledge base and projecting it into an emergent encyclopedia, SciencePedia. Our pipeline operationalizes an endpoint-driven, reductionist strategy: a Socratic agent, guided by a curriculum of around 200 courses, generates approximately 3 million first-principles questions. To ensure high fidelity, multiple independent solver models generate LCoTs, which are then rigorously filtered by prompt sanitization and cross-model answer consensus, retaining only those with verifiable endpoints. This verified corpus powers the Brainstorm Search Engine, which performs inverse knowledge search -- retrieving diverse, first-principles derivations that culminate in a target concept. This engine, in turn, feeds the Plato synthesizer, which narrates these verified chains into coherent articles. The initial SciencePedia comprises approximately 200,000 fine-grained entries spanning mathematics, physics, chemistry, biology, engineering, and computation. In evaluations across six disciplines, Plato-synthesized articles (conditioned on retrieved LCoTs) exhibit substantially higher knowledge-point density and significantly lower factual error rates than an equally-prompted baseline without retrieval (as judged by an external LLM). Built on this verifiable LCoT knowledge base, this reasoning-centric approach enables trustworthy, cross-domain scientific synthesis at scale and establishes the foundation for an ever-expanding encyclopedia.
A multi-scale sampling method for accurate and robust deep neural network to predict combustion chemical kinetics
Jan 09, 2022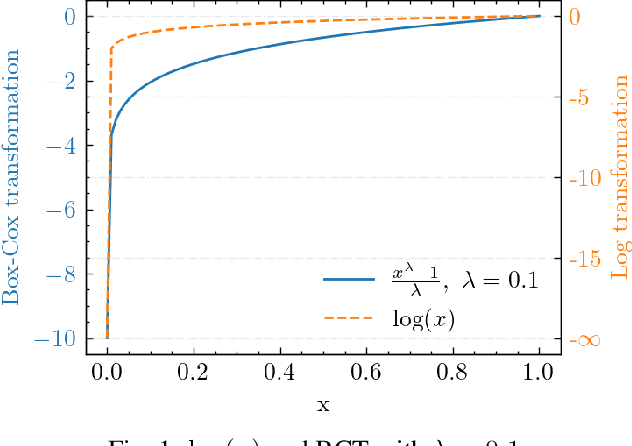
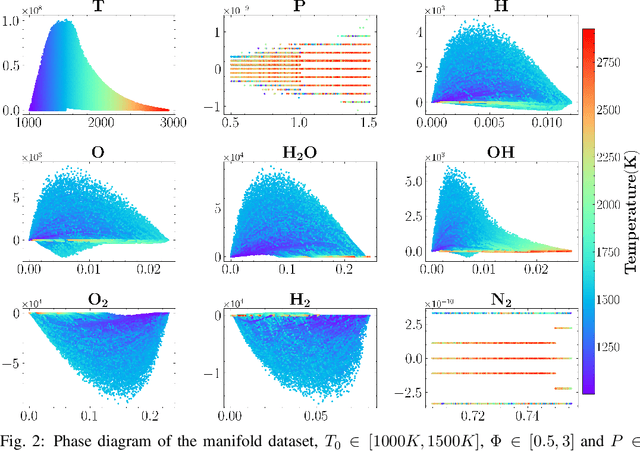
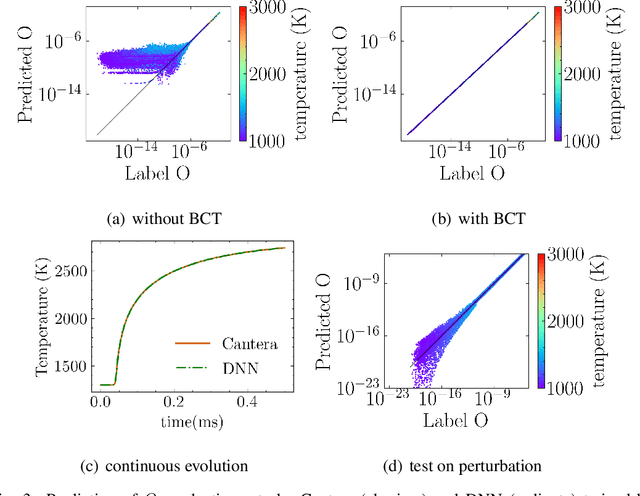
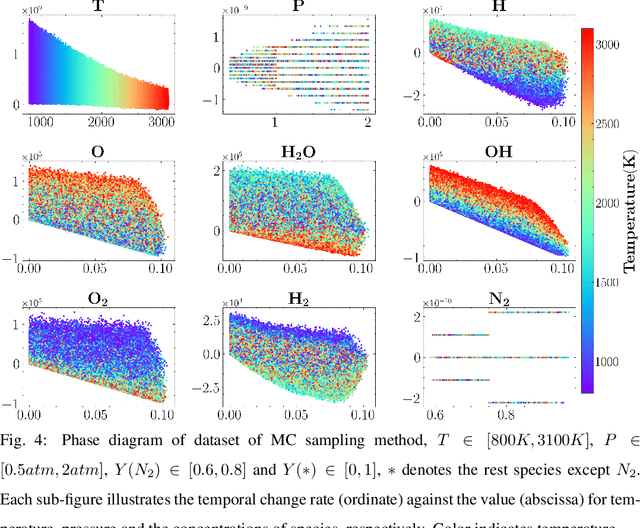
Machine learning has long been considered as a black box for predicting combustion chemical kinetics due to the extremely large number of parameters and the lack of evaluation standards and reproducibility. The current work aims to understand two basic questions regarding the deep neural network (DNN) method: what data the DNN needs and how general the DNN method can be. Sampling and preprocessing determine the DNN training dataset, further affect DNN prediction ability. The current work proposes using Box-Cox transformation (BCT) to preprocess the combustion data. In addition, this work compares different sampling methods with or without preprocessing, including the Monte Carlo method, manifold sampling, generative neural network method (cycle-GAN), and newly-proposed multi-scale sampling. Our results reveal that the DNN trained by the manifold data can capture the chemical kinetics in limited configurations but cannot remain robust toward perturbation, which is inevitable for the DNN coupled with the flow field. The Monte Carlo and cycle-GAN samplings can cover a wider phase space but fail to capture small-scale intermediate species, producing poor prediction results. A three-hidden-layer DNN, based on the multi-scale method without specific flame simulation data, allows predicting chemical kinetics in various scenarios and being stable during the temporal evolutions. This single DNN is readily implemented with several CFD codes and validated in various combustors, including (1). zero-dimensional autoignition, (2). one-dimensional freely propagating flame, (3). two-dimensional jet flame with triple-flame structure, and (4). three-dimensional turbulent lifted flames. The results demonstrate the satisfying accuracy and generalization ability of the pre-trained DNN. The Fortran and Python versions of DNN and example code are attached in the supplementary for reproducibility.
A deep learning-based model reduction method for simplifying chemical kinetics
Jan 07, 2022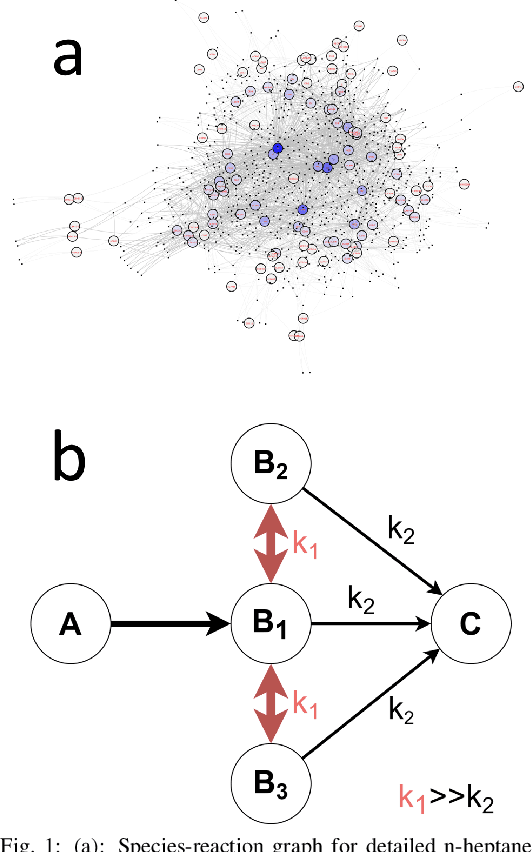
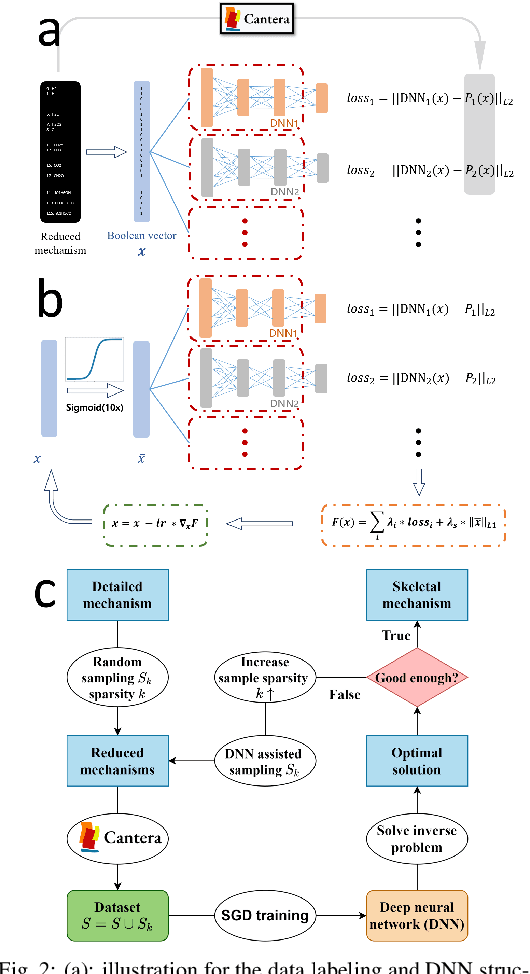
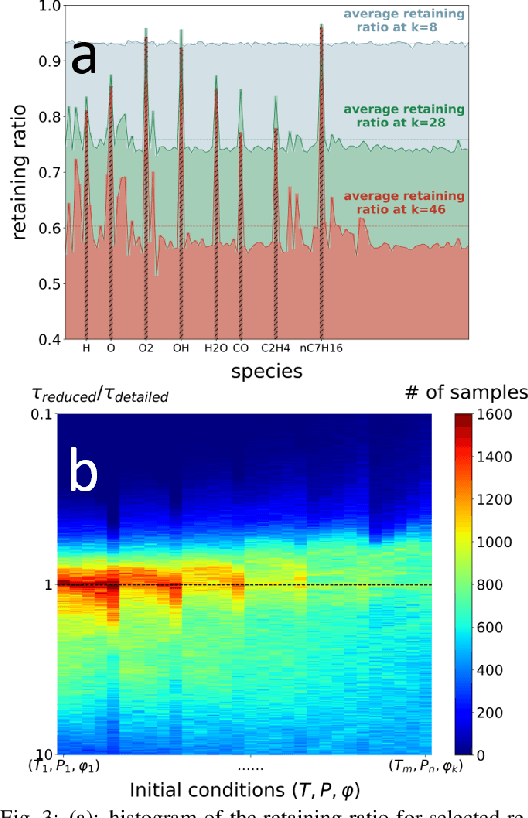
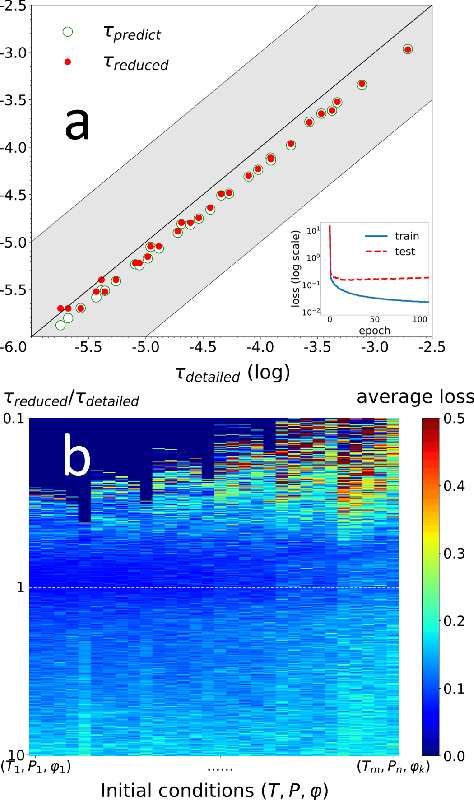
A deep learning-based model reduction (DeePMR) method for simplifying chemical kinetics is proposed and validated using high-temperature auto-ignitions, perfectly stirred reactors (PSR), and one-dimensional freely propagating flames of n-heptane/air mixtures. The mechanism reduction is modeled as an optimization problem on Boolean space, where a Boolean vector, each entry corresponding to a species, represents a reduced mechanism. The optimization goal is to minimize the reduced mechanism size given the error tolerance of a group of pre-selected benchmark quantities. The key idea of the DeePMR is to employ a deep neural network (DNN) to formulate the objective function in the optimization problem. In order to explore high dimensional Boolean space efficiently, an iterative DNN-assisted data sampling and DNN training procedure are implemented. The results show that DNN-assistance improves sampling efficiency significantly, selecting only $10^5$ samples out of $10^{34}$ possible samples for DNN to achieve sufficient accuracy. The results demonstrate the capability of the DNN to recognize key species and reasonably predict reduced mechanism performance. The well-trained DNN guarantees the optimal reduced mechanism by solving an inverse optimization problem. By comparing ignition delay times, laminar flame speeds, temperatures in PSRs, the resulting skeletal mechanism has fewer species (45 species) but the same level of accuracy as the skeletal mechanism (56 species) obtained by the Path Flux Analysis (PFA) method. In addition, the skeletal mechanism can be further reduced to 28 species if only considering atmospheric, near-stoichiometric conditions (equivalence ratio between 0.6 and 1.2). The DeePMR provides an innovative way to perform model reduction and demonstrates the great potential of data-driven methods in the combustion area.
A deep learning-based ODE solver for chemical kinetics
Nov 24, 2020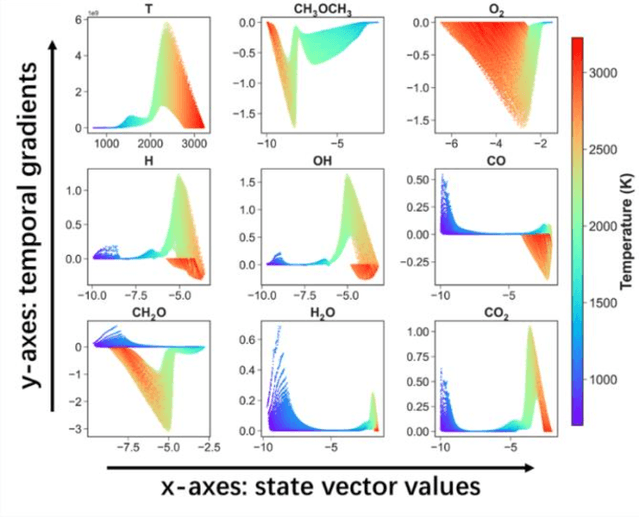
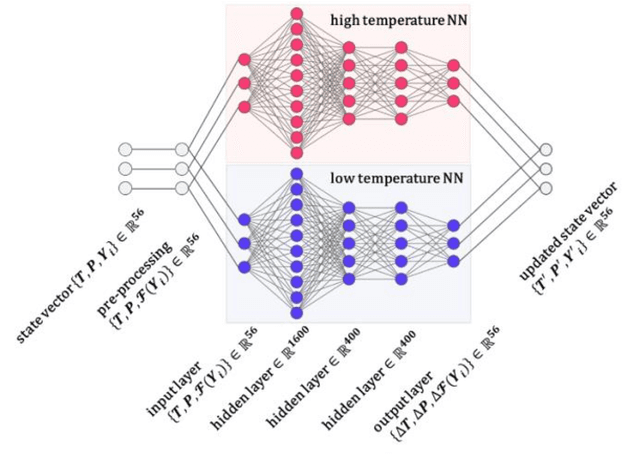
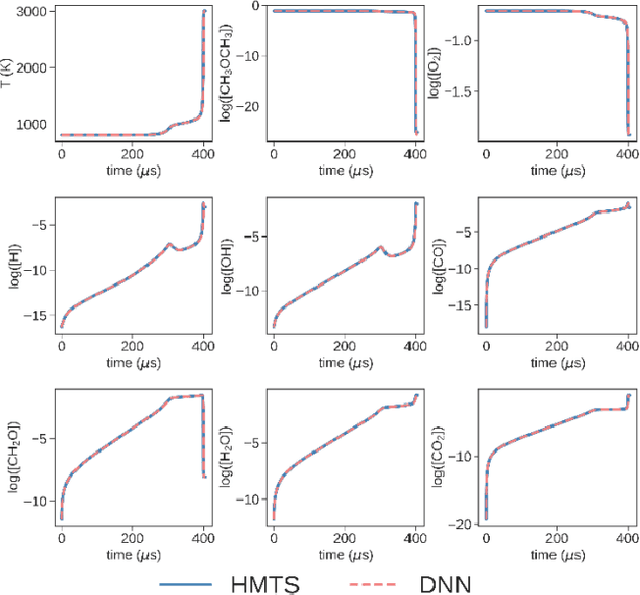
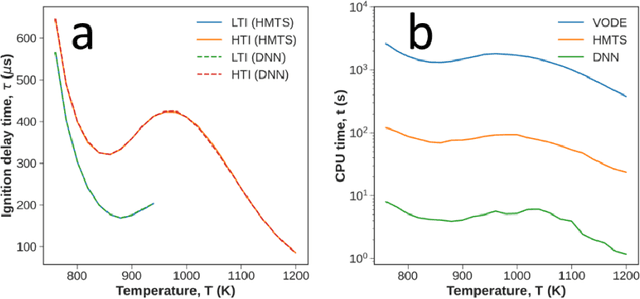
Developing efficient and accurate algorithms for chemistry integration is a challenging task due to its strong stiffness and high dimensionality. The current work presents a deep learning-based numerical method called DeepCombustion0.0 to solve stiff ordinary differential equation systems. The homogeneous autoignition of DME/air mixture, including 54 species, is adopted as an example to illustrate the validity and accuracy of the algorithm. The training and testing datasets cover a wide range of temperature, pressure, and mixture conditions between 750-1200 K, 30-50 atm, and equivalence ratio = 0.7-1.5. Both the first-stage low-temperature ignition (LTI) and the second-stage high-temperature ignition (HTI) are considered. The methodology highlights the importance of the adaptive data sampling techniques, power transform preprocessing, and binary deep neural network (DNN) design. By using the adaptive random samplings and appropriate power transforms, smooth submanifolds in the state vector phase space are observed, on which two three-layer DNNs can be appropriately trained. The neural networks are end-to-end, which predict temporal gradients of the state vectors directly. The results show that temporal evolutions predicted by DNN agree well with traditional numerical methods in all state vector dimensions, including temperature, pressure, and species concentrations. Besides, the ignition delay time differences are within 1%. At the same time, the CPU time is reduced by more than 20 times and 200 times compared with the HMTS and VODE method, respectively. The current work demonstrates the enormous potential of applying the deep learning algorithm in chemical kinetics and combustion modeling.