 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeApproximate Cross-Validation with Low-Rank Data in High Dimensions
Aug 24, 2020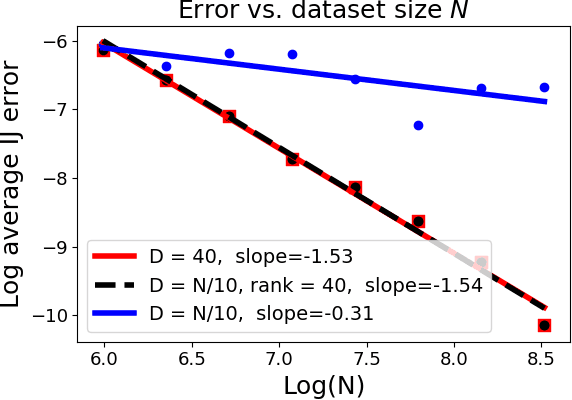
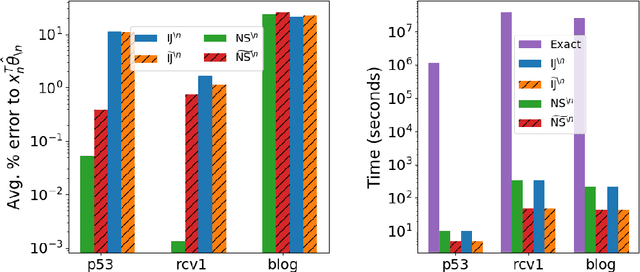
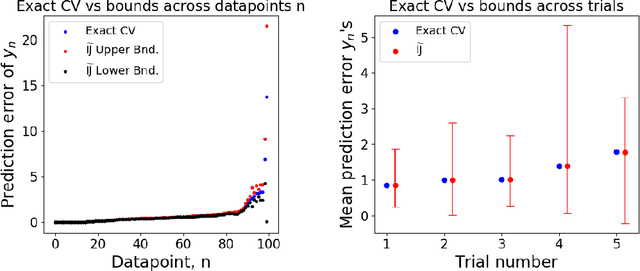
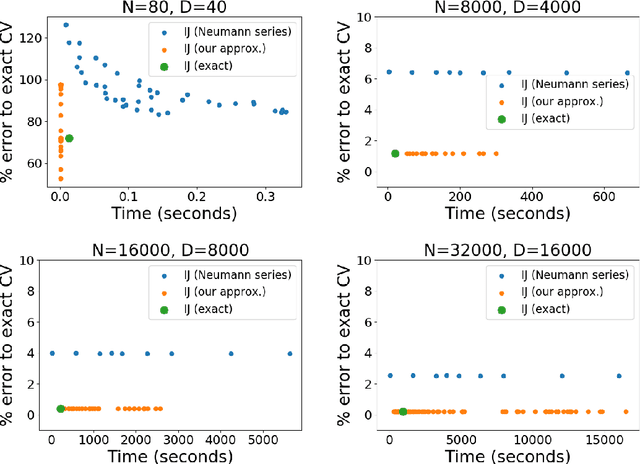
Many recent advances in machine learning are driven by a challenging trifecta: large data size $N$; high dimensions; and expensive algorithms. In this setting, cross-validation (CV) serves as an important tool for model assessment. Recent advances in approximate cross validation (ACV) provide accurate approximations to CV with only a single model fit, avoiding traditional CV's requirement for repeated runs of expensive algorithms. Unfortunately, these ACV methods can lose both speed and accuracy in high dimensions -- unless sparsity structure is present in the data. Fortunately, there is an alternative type of simplifying structure that is present in most data: approximate low rank (ALR). Guided by this observation, we develop a new algorithm for ACV that is fast and accurate in the presence of ALR data. Our first key insight is that the Hessian matrix -- whose inverse forms the computational bottleneck of existing ACV methods -- is ALR. We show that, despite our use of the \emph{inverse} Hessian, a low-rank approximation using the largest (rather than the smallest) matrix eigenvalues enables fast, reliable ACV. Our second key insight is that, in the presence of ALR data, error in existing ACV methods roughly grows with the (approximate, low) rank rather than with the (full, high) dimension. These insights allow us to prove theoretical guarantees on the quality of our proposed algorithm -- along with fast-to-compute upper bounds on its error. We demonstrate the speed and accuracy of our method, as well as the usefulness of our bounds, on a range of real and simulated data sets.
Finite mixture models are typically inconsistent for the number of components
Jul 08, 2020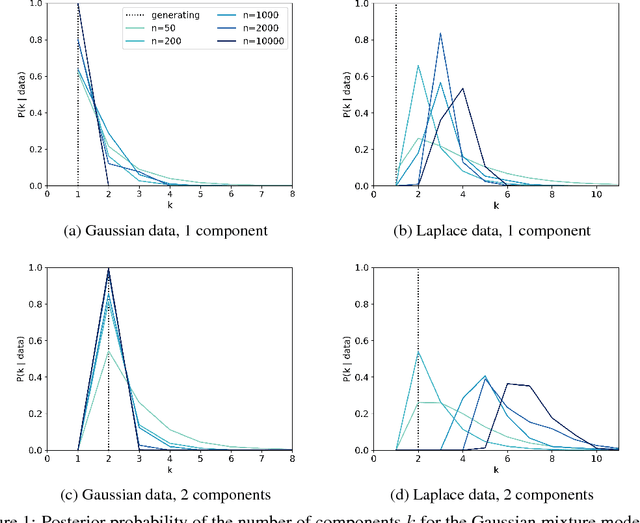
Scientists and engineers are often interested in learning the number of subpopulations (or components) present in a data set. Practitioners commonly use a Dirichlet process mixture model (DPMM) for this purpose; in particular, they count the number of clusters---i.e. components containing at least one data point---in the DPMM posterior. But Miller and Harrison (2013) warn that the DPMM cluster-count posterior is severely inconsistent for the number of latent components when the data are truly generated from a finite mixture; that is, the cluster-count posterior probability on the true generating number of components goes to zero in the limit of infinite data. A potential alternative is to use a finite mixture model (FMM) with a prior on the number of components. Past work has shown the resulting FMM component-count posterior is consistent. But existing results crucially depend on the assumption that the component likelihoods are perfectly specified. In practice, this assumption is unrealistic, and empirical evidence (Miller and Dunson, 2019) suggests that the FMM posterior on the number of components is sensitive to the likelihood choice. In this paper, we add rigor to data-analysis folk wisdom by proving that under even the slightest model misspecification, the FMM posterior on the number of components is ultraseverely inconsistent: for any finite $k \in \mathbb{N}$, the posterior probability that the number of components is $k$ converges to 0 in the limit of infinite data. We illustrate practical consequences of our theory on simulated and real data sets.
Approximate Cross-Validation for Structured Models
Jun 23, 2020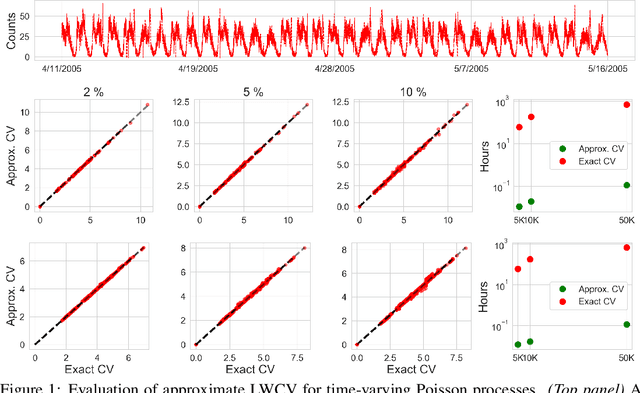
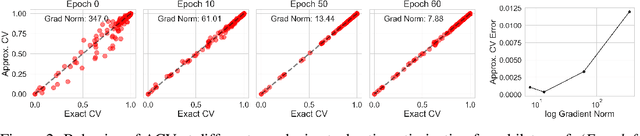
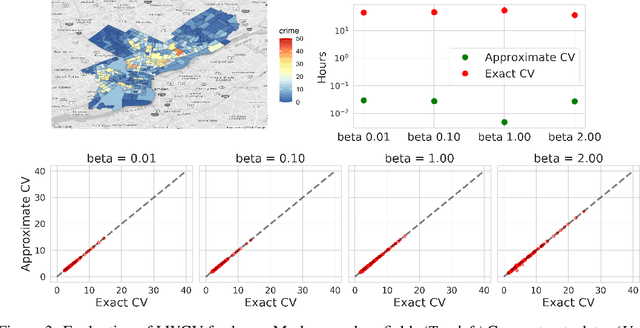
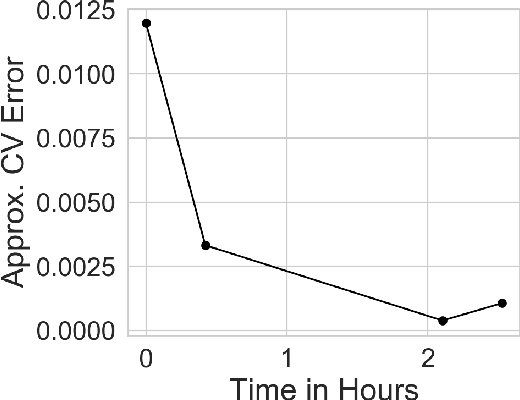
Many modern data analyses benefit from explicitly modeling dependence structure in data -- such as measurements across time or space, ordered words in a sentence, or genes in a genome. Cross-validation is the gold standard to evaluate these analyses but can be prohibitively slow due to the need to re-run already-expensive learning algorithms many times. Previous work has shown approximate cross-validation (ACV) methods provide a fast and provably accurate alternative in the setting of empirical risk minimization. But this existing ACV work is restricted to simpler models by the assumptions that (i) data are independent and (ii) an exact initial model fit is available. In structured data analyses, (i) is always untrue, and (ii) is often untrue. In the present work, we address (i) by extending ACV to models with dependence structure. To address (ii), we verify -- both theoretically and empirically -- that ACV quality deteriorates smoothly with noise in the initial fit. We demonstrate the accuracy and computational benefits of our proposed methods on a diverse set of real-world applications.
Practical Posterior Error Bounds from Variational Objectives
Oct 31, 2019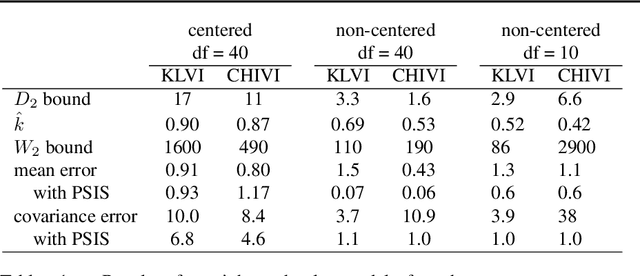
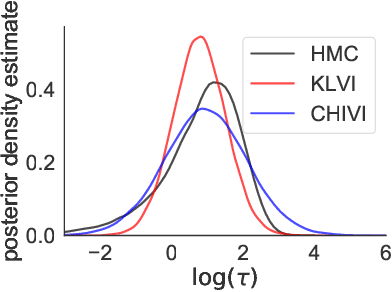
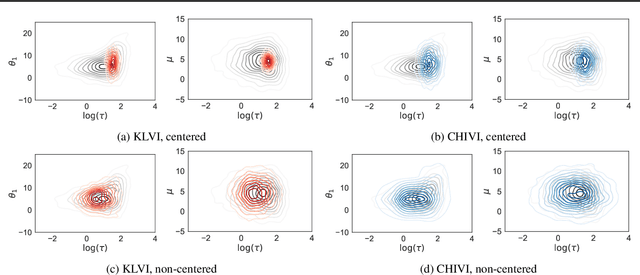
Variational inference has become an increasingly attractive fast alternative to Markov chain Monte Carlo methods for approximate Bayesian inference. However, a major obstacle to the widespread use of variational methods is the lack of post-hoc accuracy measures that are both theoretically justified and computationally efficient. In this paper, we provide rigorous bounds on the error of posterior mean and uncertainty estimates that arise from full-distribution approximations, as in variational inference. Our bounds are widely applicable as they require only that the approximating and exact posteriors have polynomial moments. Our bounds are computationally efficient for variational inference in that they require only standard values from variational objectives, straightforward analytic calculations, and simple Monte Carlo estimates. We show that our analysis naturally leads to a new and improved workflow for variational inference. Finally, we demonstrate the utility of our proposed workflow and error bounds on a real-data example with a widely used multilevel hierarchical model.
A Higher-Order Swiss Army Infinitesimal Jackknife
Jul 28, 2019
Cross validation (CV) and the bootstrap are ubiquitous model-agnostic tools for assessing the error or variability of machine learning and statistical estimators. However, these methods require repeatedly re-fitting the model with different weighted versions of the original dataset, which can be prohibitively time-consuming. For sufficiently regular optimization problems the optimum depends smoothly on the data weights, and so the process of repeatedly re-fitting can be approximated with a Taylor series that can be often evaluated relatively quickly. The first-order approximation is known as the "infinitesimal jackknife" in the statistics literature and has been the subject of recent interest in machine learning for approximate CV. In this work, we consider high-order approximations, which we call the "higher-order infinitesimal jackknife" (HOIJ). Under mild regularity conditions, we provide a simple recursive procedure to compute approximations of all orders with finite-sample accuracy bounds. Additionally, we show that the HOIJ can be efficiently computed even in high dimensions using forward-mode automatic differentiation. We show that a linear approximation with bootstrap weights approximation is equivalent to those provided by asymptotic normal approximations. Consequently, the HOIJ opens up the possibility of enjoying higher-order accuracy properties of the bootstrap using local approximations. Consistency of the HOIJ for leave-one-out CV under different asymptotic regimes follows as corollaries from our finite-sample bounds under additional regularity assumptions. The generality of the computation and bounds motivate the name "higher-order Swiss Army infinitesimal jackknife."
Sparse Approximate Cross-Validation for High-Dimensional GLMs
May 31, 2019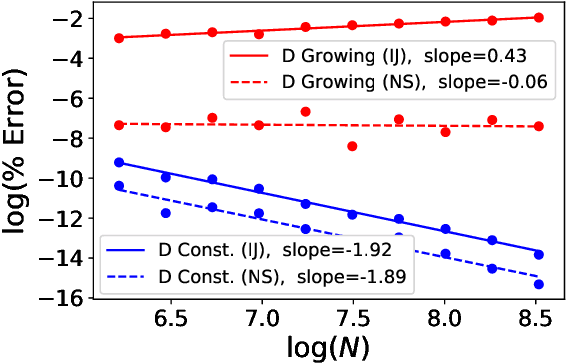
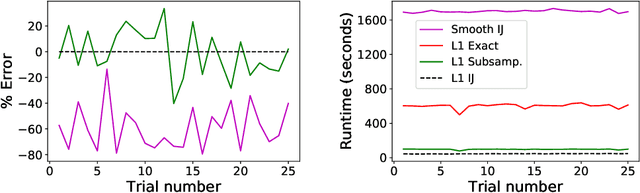
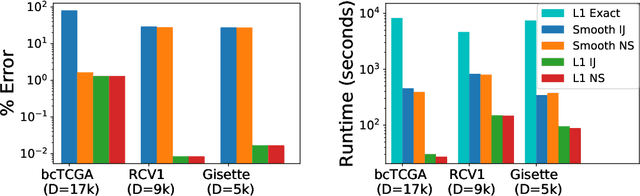
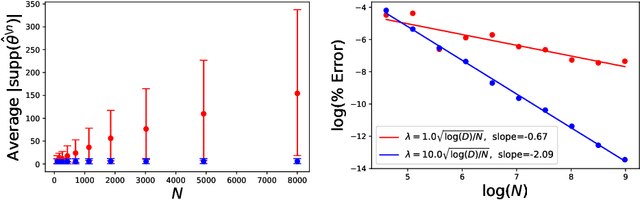
Leave-one-out cross validation (LOOCV) can be particularly accurate among CV variants for estimating out-of-sample error. Unfortunately, LOOCV requires re-fitting a model $N$ times for a dataset of size $N$. To avoid this prohibitive computational expense, a number of authors have proposed approximations to LOOCV. These approximations work well when the unknown parameter is of small, fixed dimension but suffer in high dimensions; they incur a running time roughly cubic in the dimension, and, in fact, we show their accuracy significantly deteriorates in high dimensions. We demonstrate that these difficulties can be surmounted in $\ell_1$-regularized generalized linear models when we assume that the unknown parameter, while high dimensional, has a small support. In particular, we show that, under interpretable conditions, the support of the recovered parameter does not change as each datapoint is left out. This result implies that the previously proposed heuristic of only approximating CV along the support of the recovered parameter has running time and error that scale with the (small) support size even when the full dimension is large. Experiments on synthetic and real data support the accuracy of our approximations.
LR-GLM: High-Dimensional Bayesian Inference Using Low-Rank Data Approximations
May 17, 2019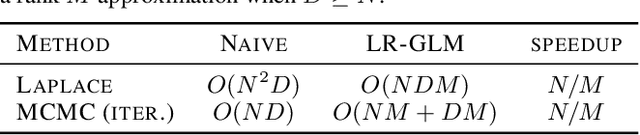
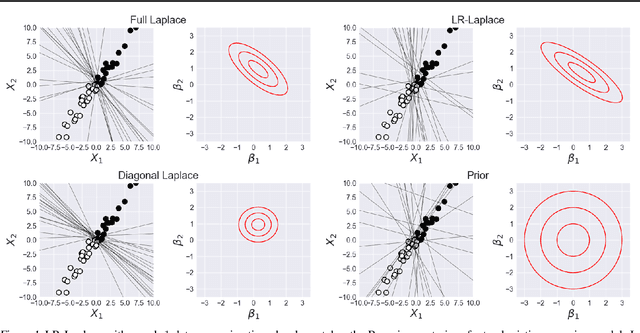
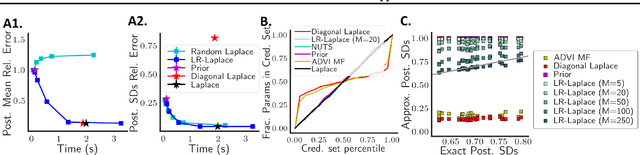
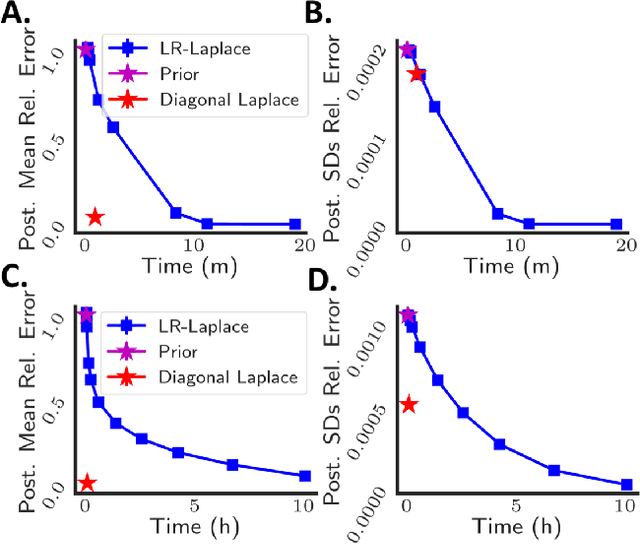
Due to the ease of modern data collection, applied statisticians often have access to a large set of covariates that they wish to relate to some observed outcome. Generalized linear models (GLMs) offer a particularly interpretable framework for such an analysis. In these high-dimensional problems, the number of covariates is often large relative to the number of observations, so we face non-trivial inferential uncertainty; a Bayesian approach allows coherent quantification of this uncertainty. Unfortunately, existing methods for Bayesian inference in GLMs require running times roughly cubic in parameter dimension, and so are limited to settings with at most tens of thousand parameters. We propose to reduce time and memory costs with a low-rank approximation of the data in an approach we call LR-GLM. When used with the Laplace approximation or Markov chain Monte Carlo, LR-GLM provides a full Bayesian posterior approximation and admits running times reduced by a full factor of the parameter dimension. We rigorously establish the quality of our approximation and show how the choice of rank allows a tunable computational-statistical trade-off. Experiments support our theory and demonstrate the efficacy of LR-GLM on real large-scale datasets.
The Kernel Interaction Trick: Fast Bayesian Discovery of Pairwise Interactions in High Dimensions
May 16, 2019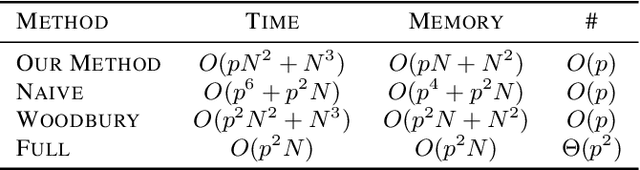
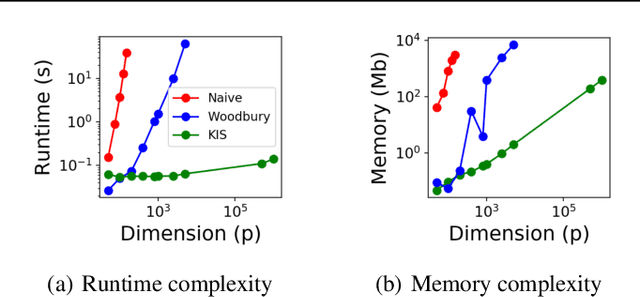
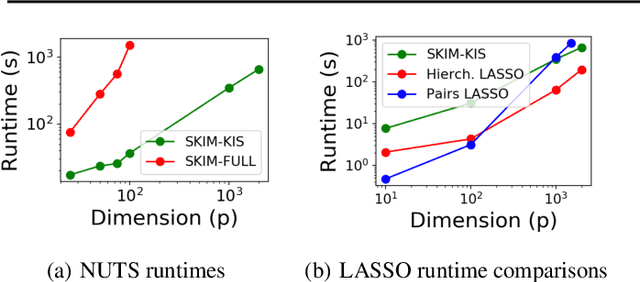
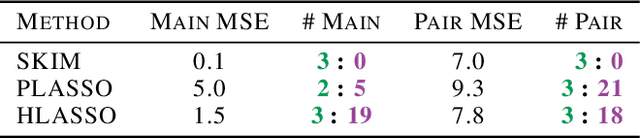
Discovering interaction effects on a response of interest is a fundamental problem faced in biology, medicine, economics, and many other scientific disciplines. In theory, Bayesian methods for discovering pairwise interactions enjoy many benefits such as coherent uncertainty quantification, the ability to incorporate background knowledge, and desirable shrinkage properties. In practice, however, Bayesian methods are often computationally intractable for even moderate-dimensional problems. Our key insight is that many hierarchical models of practical interest admit a particular Gaussian process (GP) representation; the GP allows us to capture the posterior with a vector of O(p) kernel hyper-parameters rather than O(p^2) interactions and main effects. With the implicit representation, we can run Markov chain Monte Carlo (MCMC) over model hyper-parameters in time and memory linear in p per iteration. We focus on sparsity-inducing models and show on datasets with a variety of covariate behaviors that our method: (1) reduces runtime by orders of magnitude over naive applications of MCMC, (2) provides lower Type I and Type II error relative to state-of-the-art LASSO-based approaches, and (3) offers improved computational scaling in high dimensions relative to existing Bayesian and LASSO-based approaches.
Reconstructing probabilistic trees of cellular differentiation from single-cell RNA-seq data
Nov 28, 2018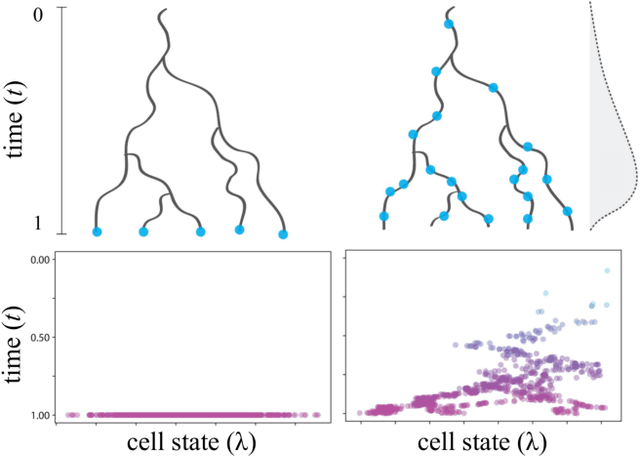
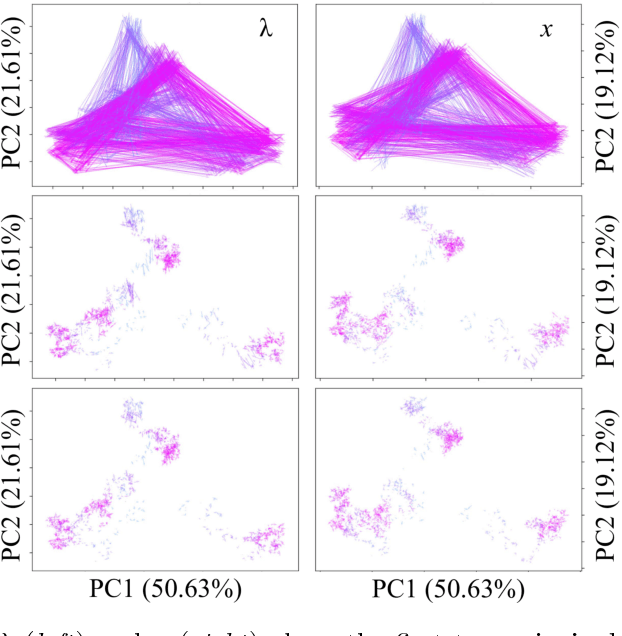
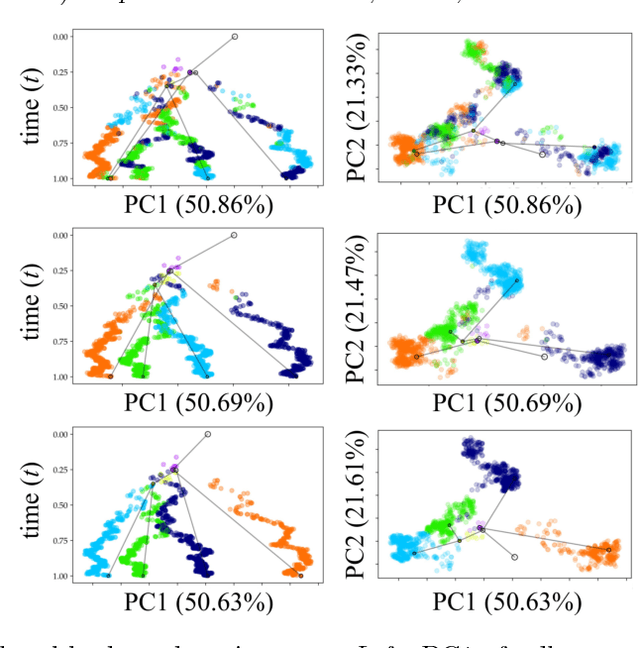
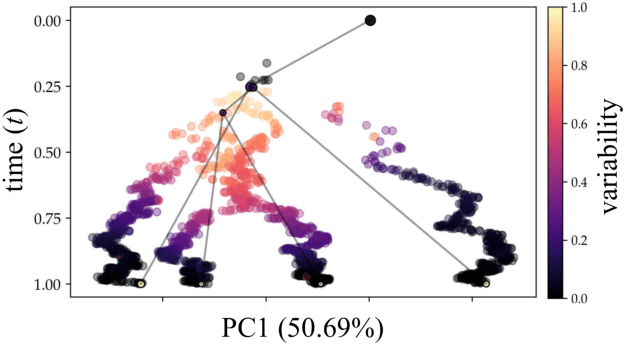
Until recently, transcriptomics was limited to bulk RNA sequencing, obscuring the underlying expression patterns of individual cells in favor of a global average. Thanks to technological advances, we can now profile gene expression across thousands or millions of individual cells in parallel. This new type of data has led to the intriguing discovery that individual cell profiles can reflect the imprint of time or dynamic processes. However, synthesizing this information to reconstruct dynamic biological phenomena from data that are noisy, heterogenous, and sparse---and from processes that may unfold asynchronously---poses a complex computational and statistical challenge. Here, we develop a full generative model for probabilistically reconstructing trees of cellular differentiation from single-cell RNA-seq data. Specifically, we extend the framework of the classical Dirichlet diffusion tree to simultaneously infer branch topology and latent cell states along continuous trajectories over the full tree. In tandem, we construct a novel Markov chain Monte Carlo sampler that interleaves Metropolis-Hastings and message passing to leverage model structure for efficient inference. Finally, we demonstrate that these techniques can recover latent trajectories from simulated single-cell transcriptomes. While this work is motivated by cellular differentiation, we derive a tractable model that provides flexible densities for any data (coupled with an appropriate noise model) that arise from continuous evolution along a latent nonparametric tree.
Data-dependent compression of random features for large-scale kernel approximation
Oct 09, 2018
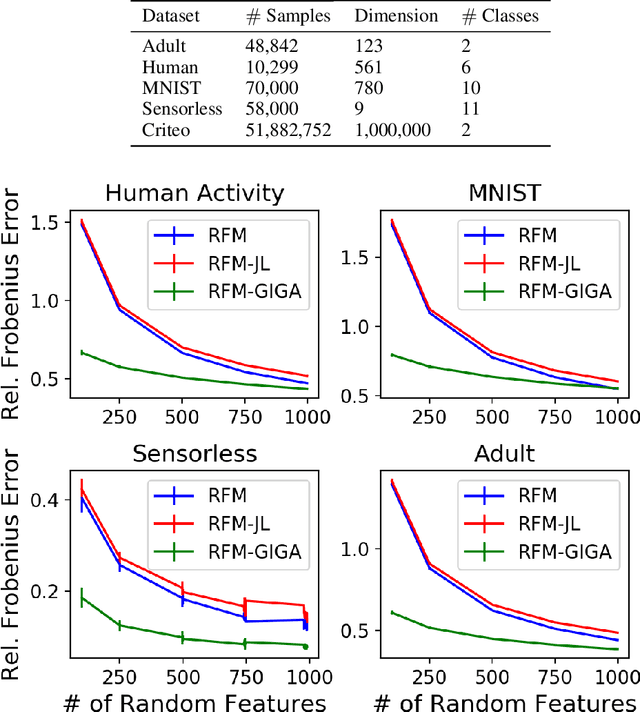

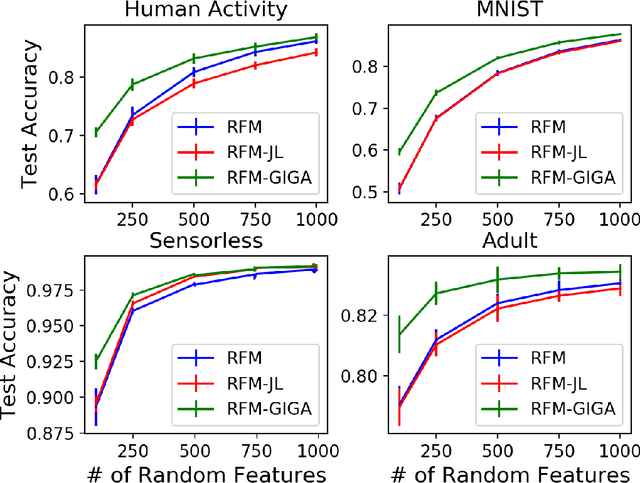
Kernel methods offer the flexibility to learn complex relationships in modern, large data sets while enjoying strong theoretical guarantees on quality. Unfortunately, these methods typically require cubic running time in the data set size, a prohibitive cost in the large-data setting. Random feature maps (RFMs) and the Nystrom method both consider low-rank approximations to the kernel matrix as a potential solution. But, in order to achieve desirable theoretical guarantees, the former may require a prohibitively large number of features J+, and the latter may be prohibitively expensive for high-dimensional problems. We propose to combine the simplicity and generality of RFMs with a data-dependent feature selection scheme to achieve desirable theoretical approximation properties of Nystrom with just O(log J+) features. Our key insight is to begin with a large set of random features, then reduce them to a small number of weighted features in a data-dependent, computationally efficient way, while preserving the statistical guarantees of using the original large set of features. We demonstrate the efficacy of our method with theory and experiments--including on a data set with over 50 million observations. In particular, we show that our method achieves small kernel matrix approximation error and better test set accuracy with provably fewer random features than state- of-the-art methods.