 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTerraBind: Fast and Accurate Binding Affinity Prediction through Coarse Structural Representations
Feb 08, 2026We present TerraBind, a foundation model for protein-ligand structure and binding affinity prediction that achieves 26-fold faster inference than state-of-the-art methods while improving affinity prediction accuracy by $\sim$20\%. Current deep learning approaches to structure-based drug design rely on expensive all-atom diffusion to generate 3D coordinates, creating inference bottlenecks that render large-scale compound screening computationally intractable. We challenge this paradigm with a critical hypothesis: full all-atom resolution is unnecessary for accurate small molecule pose and binding affinity prediction. TerraBind tests this hypothesis through a coarse pocket-level representation (protein C$_β$ atoms and ligand heavy atoms only) within a multimodal architecture combining COATI-3 molecular encodings and ESM-2 protein embeddings that learns rich structural representations, which are used in a diffusion-free optimization module for pose generation and a binding affinity likelihood prediction module. On structure prediction benchmarks (FoldBench, PoseBusters, Runs N' Poses), TerraBind matches diffusion-based baselines in ligand pose accuracy. Crucially, TerraBind outperforms Boltz-2 by $\sim$20\% in Pearson correlation for binding affinity prediction on both a public benchmark (CASP16) and a diverse proprietary dataset (18 biochemical/cell assays). We show that the affinity prediction module also provides well-calibrated affinity uncertainty estimates, addressing a critical gap in reliable compound prioritization for drug discovery. Furthermore, this module enables a continual learning framework and a hedged batch selection strategy that, in simulated drug discovery cycles, achieves 6$\times$ greater affinity improvement of selected molecules over greedy-based approaches.
Pretrained Joint Predictions for Scalable Batch Bayesian Optimization of Molecular Designs
Nov 14, 2025Batched synthesis and testing of molecular designs is the key bottleneck of drug development. There has been great interest in leveraging biomolecular foundation models as surrogates to accelerate this process. In this work, we show how to obtain scalable probabilistic surrogates of binding affinity for use in Batch Bayesian Optimization (Batch BO). This demands parallel acquisition functions that hedge between designs and the ability to rapidly sample from a joint predictive density to approximate them. Through the framework of Epistemic Neural Networks (ENNs), we obtain scalable joint predictive distributions of binding affinity on top of representations taken from large structure-informed models. Key to this work is an investigation into the importance of prior networks in ENNs and how to pretrain them on synthetic data to improve downstream performance in Batch BO. Their utility is demonstrated by rediscovering known potent EGFR inhibitors on a semi-synthetic benchmark in up to 5x fewer iterations, as well as potent inhibitors from a real-world small-molecule library in up to 10x fewer iterations, offering a promising solution for large-scale drug discovery applications.
Generalizability of density functionals learned from differentiable programming on weakly correlated spin-polarized systems
Oct 28, 2021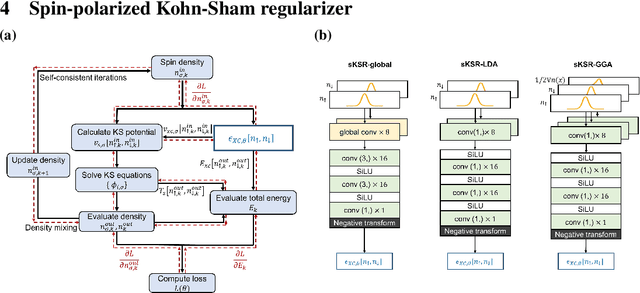

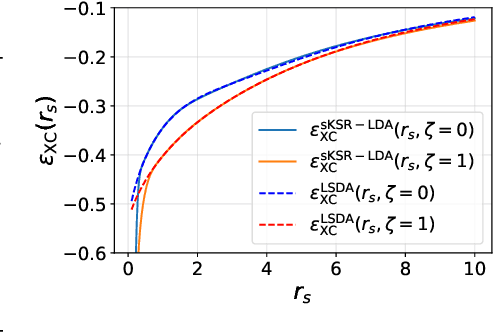
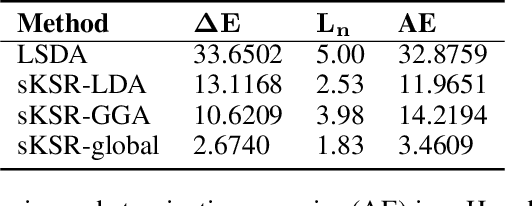
Kohn-Sham regularizer (KSR) is a machine learning approach that optimizes a physics-informed exchange-correlation functional within a differentiable Kohn-Sham density functional theory framework. We evaluate the generalizability of KSR by training on atomic systems and testing on molecules at equilibrium. We propose a spin-polarized version of KSR with local, semilocal, and nonlocal approximations for the exchange-correlation functional. The generalization error from our semilocal approximation is comparable to other differentiable approaches. Our nonlocal functional outperforms any existing machine learning functionals by predicting the ground-state energies of the test systems with a mean absolute error of 2.7 milli-Hartrees.
Kohn-Sham equations as regularizer: building prior knowledge into machine-learned physics
Sep 17, 2020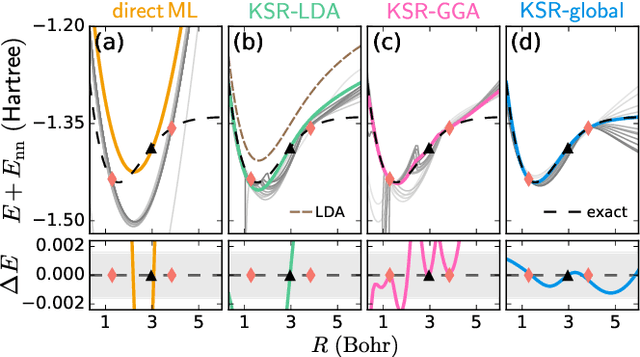
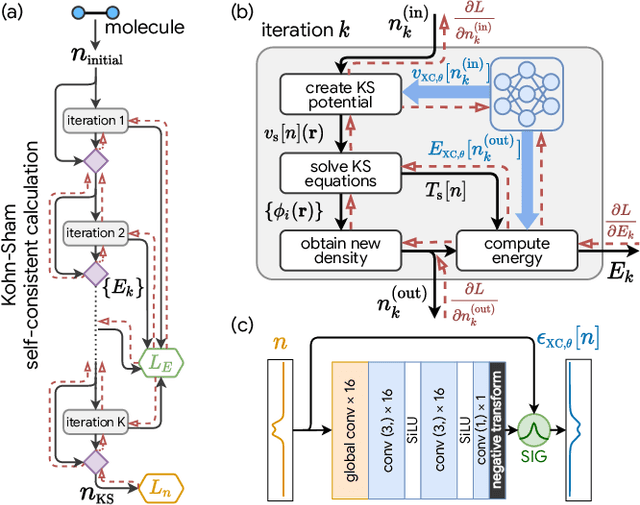
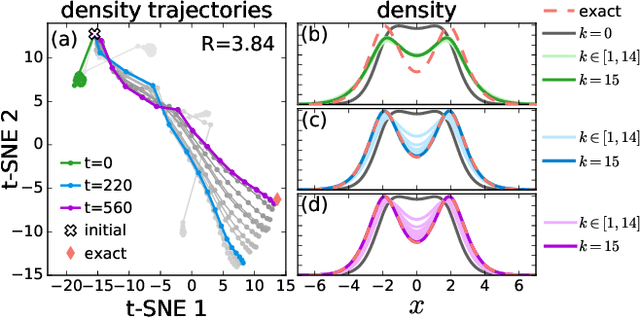
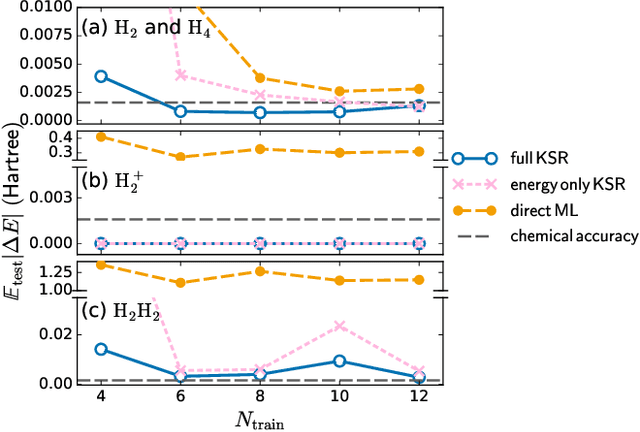
Including prior knowledge is important for effective machine learning models in physics, and is usually achieved by explicitly adding loss terms or constraints on model architectures. Prior knowledge embedded in the physics computation itself rarely draws attention. We show that solving the Kohn-Sham equations when training neural networks for the exchange-correlation functional provides an implicit regularization that greatly improves generalization. Two separations suffice for learning the entire one-dimensional H$_2$ dissociation curve within chemical accuracy, including the strongly correlated region. Our models also generalize to unseen types of molecules and overcome self-interaction error.