 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePretrained Joint Predictions for Scalable Batch Bayesian Optimization of Molecular Designs
Nov 14, 2025Batched synthesis and testing of molecular designs is the key bottleneck of drug development. There has been great interest in leveraging biomolecular foundation models as surrogates to accelerate this process. In this work, we show how to obtain scalable probabilistic surrogates of binding affinity for use in Batch Bayesian Optimization (Batch BO). This demands parallel acquisition functions that hedge between designs and the ability to rapidly sample from a joint predictive density to approximate them. Through the framework of Epistemic Neural Networks (ENNs), we obtain scalable joint predictive distributions of binding affinity on top of representations taken from large structure-informed models. Key to this work is an investigation into the importance of prior networks in ENNs and how to pretrain them on synthetic data to improve downstream performance in Batch BO. Their utility is demonstrated by rediscovering known potent EGFR inhibitors on a semi-synthetic benchmark in up to 5x fewer iterations, as well as potent inhibitors from a real-world small-molecule library in up to 10x fewer iterations, offering a promising solution for large-scale drug discovery applications.
Machine learning and AI-based approaches for bioactive ligand discovery and GPCR-ligand recognition
Jan 22, 2020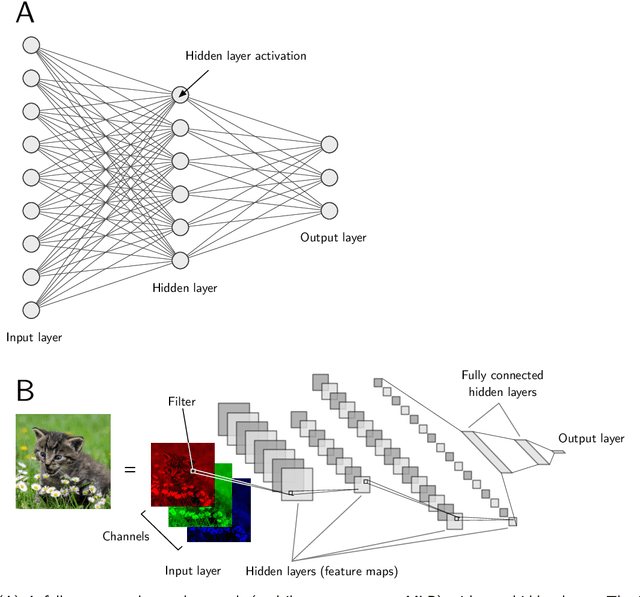
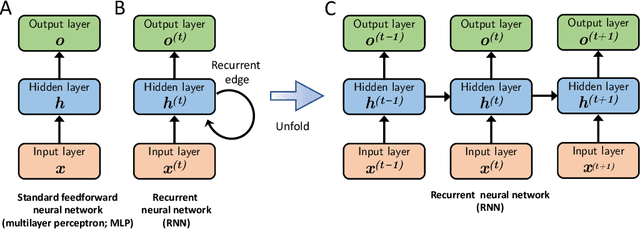
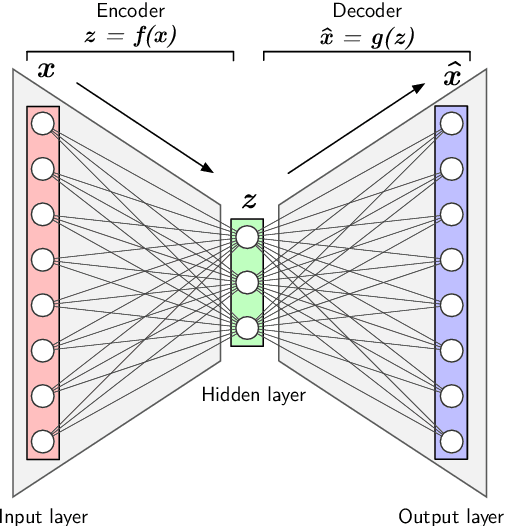
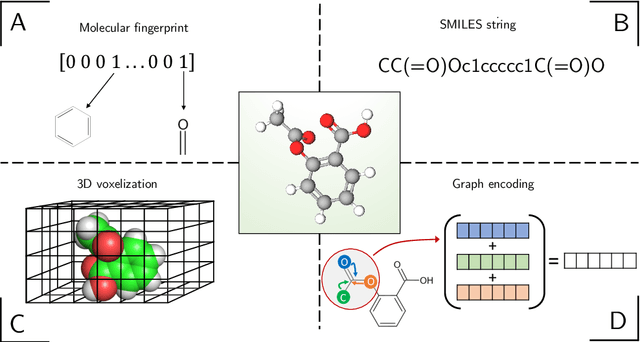
In the last decade, machine learning and artificial intelligence applications have received a significant boost in performance and attention in both academic research and industry. The success behind most of the recent state-of-the-art methods can be attributed to the latest developments in deep learning. When applied to various scientific domains that are concerned with the processing of non-tabular data, for example, image or text, deep learning has been shown to outperform not only conventional machine learning but also highly specialized tools developed by domain experts. This review aims to summarize AI-based research for GPCR bioactive ligand discovery with a particular focus on the most recent achievements and research trends. To make this article accessible to a broad audience of computational scientists, we provide instructive explanations of the underlying methodology, including overviews of the most commonly used deep learning architectures and feature representations of molecular data. We highlight the latest AI-based research that has led to the successful discovery of GPCR bioactive ligands. However, an equal focus of this review is on the discussion of machine learning-based technology that has been applied to ligand discovery in general and has the potential to pave the way for successful GPCR bioactive ligand discovery in the future. This review concludes with a brief outlook highlighting the recent research trends in deep learning, such as active learning and semi-supervised learning, which have great potential for advancing bioactive ligand discovery.