 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHistoAtlas: A Pan-Cancer Morphology Atlas Linking Histomics to Molecular Programs and Clinical Outcomes
Mar 17, 2026We present HistoAtlas, a pan-cancer computational atlas that extracts 38 interpretable histomic features from 6,745 diagnostic H&E slides across 21 TCGA cancer types and systematically links every feature to survival, gene expression, somatic mutations, and immune subtypes. All associations are covariate-adjusted, multiple-testing corrected, and classified into evidence-strength tiers. The atlas recovers known biology, from immune infiltration and prognosis to proliferation and kinase signaling, while uncovering compartment-specific immune signals and morphological subtypes with divergent outcomes. Every result is spatially traceable to tissue compartments and individual cells, statistically calibrated, and openly queryable. HistoAtlas enables systematic, large-scale biomarker discovery from routine H&E without specialized staining or sequencing. Data and an interactive web atlas are freely available at https://histoatlas.com .
IMILIA: interpretable multiple instance learning for inflammation prediction in IBD from H&E whole slide images
Dec 15, 2025As the therapeutic target for Inflammatory Bowel Disease (IBD) shifts toward histologic remission, the accurate assessment of microscopic inflammation has become increasingly central for evaluating disease activity and response to treatment. In this work, we introduce IMILIA (Interpretable Multiple Instance Learning for Inflammation Analysis), an end-to-end framework designed for the prediction of inflammation presence in IBD digitized slides stained with hematoxylin and eosin (H&E), followed by the automated computation of markers characterizing tissue regions driving the predictions. IMILIA is composed of an inflammation prediction module, consisting of a Multiple Instance Learning (MIL) model, and an interpretability module, divided in two blocks: HistoPLUS, for cell instance detection, segmentation and classification; and EpiSeg, for epithelium segmentation. IMILIA achieves a cross-validation ROC-AUC of 0.83 on the discovery cohort, and a ROC-AUC of 0.99 and 0.84 on two external validation cohorts. The interpretability module yields biologically consistent insights: tiles with higher predicted scores show increased densities of immune cells (lymphocytes, plasmocytes, neutrophils and eosinophils), whereas lower-scored tiles predominantly contain normal epithelial cells. Notably, these patterns were consistent across all datasets. Code and models to partially replicate the results on the public IBDColEpi dataset can be found at https://github.com/owkin/imilia.
Towards Comprehensive Cellular Characterisation of H&E slides
Aug 13, 2025Cell detection, segmentation and classification are essential for analyzing tumor microenvironments (TME) on hematoxylin and eosin (H&E) slides. Existing methods suffer from poor performance on understudied cell types (rare or not present in public datasets) and limited cross-domain generalization. To address these shortcomings, we introduce HistoPLUS, a state-of-the-art model for cell analysis, trained on a novel curated pan-cancer dataset of 108,722 nuclei covering 13 cell types. In external validation across 4 independent cohorts, HistoPLUS outperforms current state-of-the-art models in detection quality by 5.2% and overall F1 classification score by 23.7%, while using 5x fewer parameters. Notably, HistoPLUS unlocks the study of 7 understudied cell types and brings significant improvements on 8 of 13 cell types. Moreover, we show that HistoPLUS robustly transfers to two oncology indications unseen during training. To support broader TME biomarker research, we release the model weights and inference code at https://github.com/owkin/histoplus/.
Benchopt: Reproducible, efficient and collaborative optimization benchmarks
Jun 28, 2022
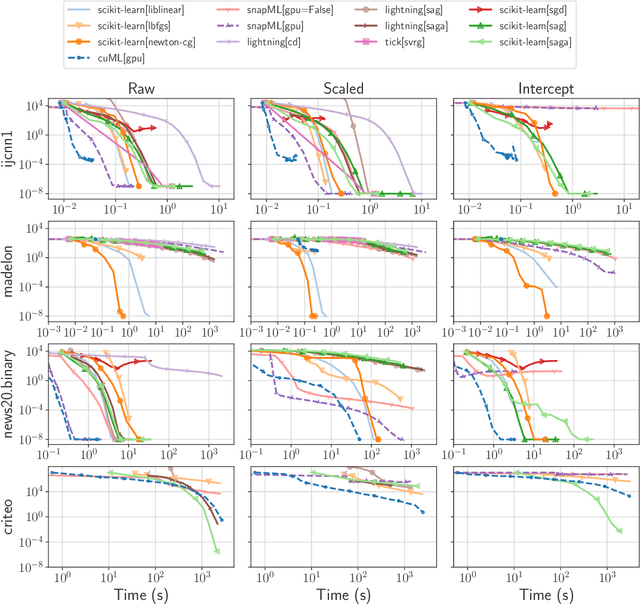
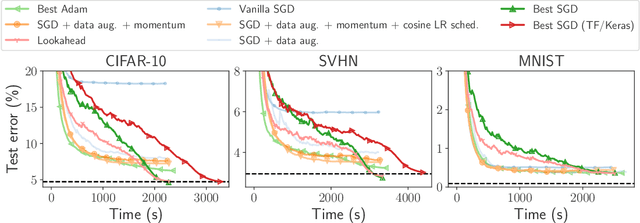
Numerical validation is at the core of machine learning research as it allows to assess the actual impact of new methods, and to confirm the agreement between theory and practice. Yet, the rapid development of the field poses several challenges: researchers are confronted with a profusion of methods to compare, limited transparency and consensus on best practices, as well as tedious re-implementation work. As a result, validation is often very partial, which can lead to wrong conclusions that slow down the progress of research. We propose Benchopt, a collaborative framework to automate, reproduce and publish optimization benchmarks in machine learning across programming languages and hardware architectures. Benchopt simplifies benchmarking for the community by providing an off-the-shelf tool for running, sharing and extending experiments. To demonstrate its broad usability, we showcase benchmarks on three standard learning tasks: $\ell_2$-regularized logistic regression, Lasso, and ResNet18 training for image classification. These benchmarks highlight key practical findings that give a more nuanced view of the state-of-the-art for these problems, showing that for practical evaluation, the devil is in the details. We hope that Benchopt will foster collaborative work in the community hence improving the reproducibility of research findings.
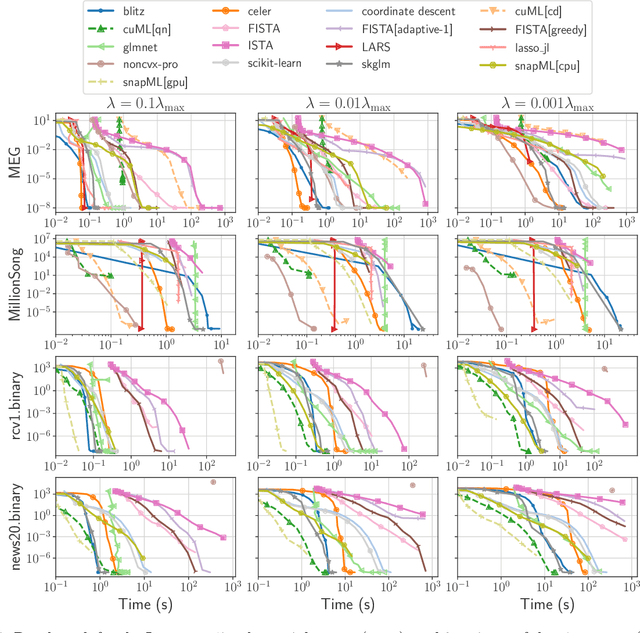
Beyond L1: Faster and Better Sparse Models with skglm
Apr 16, 2022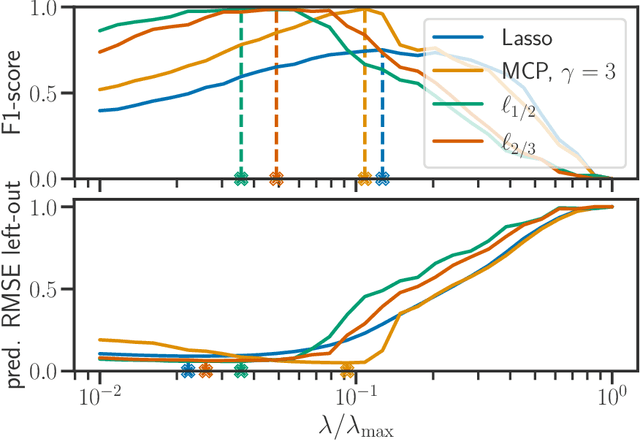
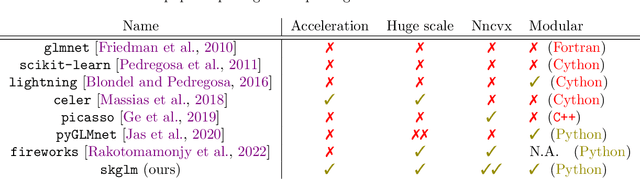
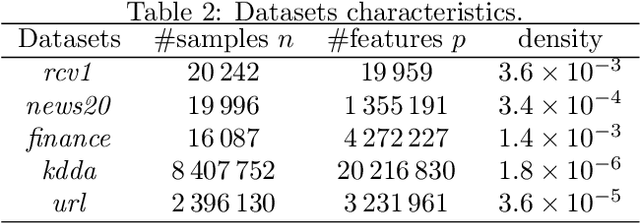
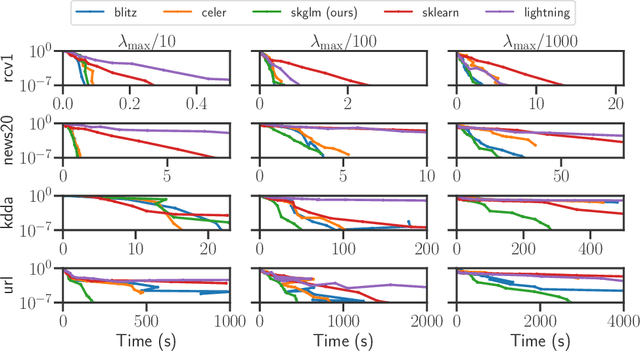
We propose a new fast algorithm to estimate any sparse generalized linear model with convex or non-convex separable penalties. Our algorithm is able to solve problems with millions of samples and features in seconds, by relying on coordinate descent, working sets and Anderson acceleration. It handles previously unaddressed models, and is extensively shown to improve state-of-art algorithms. We provide a flexible, scikit-learn compatible package, which easily handles customized datafits and penalties.