 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePoint transformer for protein structural heterogeneity analysis using CryoEM
Jan 26, 2026Structural dynamics of macromolecules is critical to their structural-function relationship. Cryogenic electron microscopy (CryoEM) provides snapshots of vitrified protein at different compositional and conformational states, and the structural heterogeneity of proteins can be characterized through computational analysis of the images. For protein systems with multiple degrees of freedom, it is still challenging to disentangle and interpret the different modes of dynamics. Here, by implementing Point Transformer, a self-attention network designed for point cloud analysis, we are able to improve the performance of heterogeneity analysis on CryoEM data, and characterize the dynamics of highly complex protein systems in a more human-interpretable way.
Unsupervised SE(3) Disentanglement for in situ Macromolecular Morphology Identification from Cryo-Electron Tomography
Jan 04, 2026Cryo-electron tomography (cryo-ET) provides direct 3D visualization of macromolecules inside the cell, enabling analysis of their in situ morphology. This morphology can be regarded as an SE(3)-invariant, denoised volumetric representation of subvolumes extracted from tomograms. Inferring morphology is therefore an inverse problem of estimating both a template morphology and its SE(3) transformation. Existing expectation-maximization based solution to this problem often misses rare but important morphologies and requires extensive manual hyperparameter tuning. Addressing this issue, we present a disentangled deep representation learning framework that separates SE(3) transformations from morphological content in the representation space. The framework includes a novel multi-choice learning module that enables this disentanglement for highly noisy cryo-ET data, and the learned morphological content is used to generate template morphologies. Experiments on simulated and real cryo-ET datasets demonstrate clear improvements over prior methods, including the discovery of previously unidentified macromolecular morphologies.
Deep learning based mixed-dimensional GMM for characterizing variability in CryoEM
Jan 25, 2021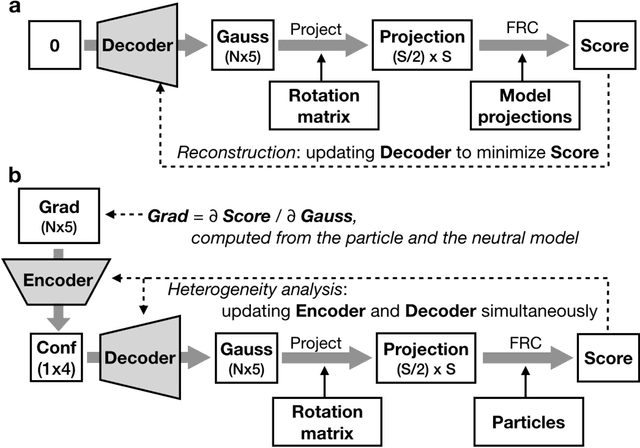

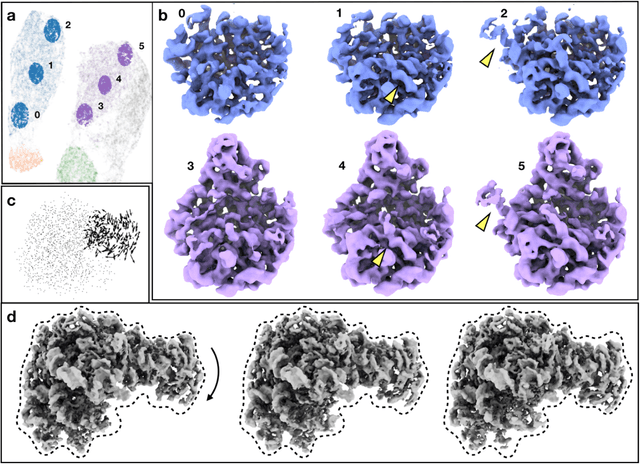
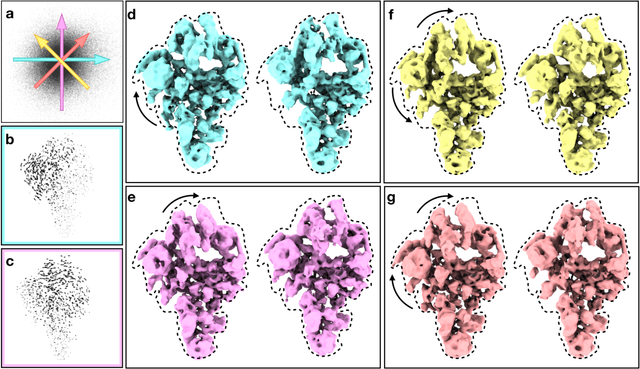
The function of most protein molecules involves structural flexibility and/or dynamic interactions with other molecules. CryoEM provides direct visualization of individual macromolecules in different conformational and compositional states. While many methods are available for classification of discrete states, characterization of continuous conformational changes or large numbers of discrete state without human supervision remains challenging. Here we present a machine learning algorithm to determine a conformational landscape for proteins or complexes using a 3-D Gaussian mixture model mapped onto 2-D particle images in known orientations. Using a deep neural network architecture, this method can automatically resolve the structural heterogeneity within the protein complex and map particles onto a small latent space describing conformational and compositional changes. This system presents a more intuitive and flexible representation than other manifold methods currently in use. We demonstrate this method on several different biomolecular systems to explore compositional and conformational changes at a range of scales.