 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-Constraint Molecular Generation using Sparsely Labelled Training Data for Localized High-Concentration Electrolyte Diluent Screening
Jan 12, 2023



Recently, machine learning methods have been used to propose molecules with desired properties, which is especially useful for exploring large chemical spaces efficiently. However, these methods rely on fully labelled training data, and are not practical in situations where molecules with multiple property constraints are required. There is often insufficient training data for all those properties from publicly available databases, especially when ab-initio simulation or experimental property data is also desired for training the conditional molecular generative model. In this work, we show how to modify a semi-supervised variational auto-encoder (SSVAE) model which only works with fully labelled and fully unlabelled molecular property training data into the ConGen model, which also works on training data that have sparsely populated labels. We evaluate ConGen's performance in generating molecules with multiple constraints when trained on a dataset combined from multiple publicly available molecule property databases, and demonstrate an example application of building the virtual chemical space for potential Lithium-ion battery localized high-concentration electrolyte (LHCE) diluents.
Protein-Ligand Complex Generator & Drug Screening via Tiered Tensor Transform
Jan 03, 2023



Accurate determination of a small molecule candidate (ligand) binding pose in its target protein pocket is important for computer-aided drug discovery. Typical rigid-body docking methods ignore the pocket flexibility of protein, while the more accurate pose generation using molecular dynamics is hindered by slow protein dynamics. We develop a tiered tensor transform (3T) algorithm to rapidly generate diverse protein-ligand complex conformations for both pose and affinity estimation in drug screening, requiring neither machine learning training nor lengthy dynamics computation, while maintaining both coarse-grain-like coordinated protein dynamics and atomistic-level details of the complex pocket. The 3T conformation structures we generate are closer to experimental co-crystal structures than those generated by docking software, and more importantly achieve significantly higher accuracy in active ligand classification than traditional ensemble docking using hundreds of experimental protein conformations. 3T structure transformation is decoupled from the system physics, making future usage in other computational scientific domains possible.
SE-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials
Jan 08, 2021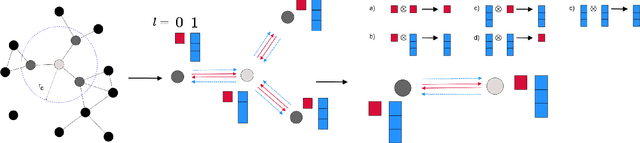
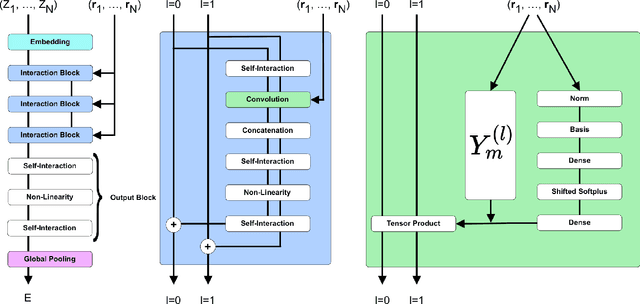
This work presents Neural Equivariant Interatomic Potentials (NequIP), a SE(3)-equivariant neural network approach for learning interatomic potentials from ab-initio calculations for molecular dynamics simulations. While most contemporary symmetry-aware models use invariant convolutions and only act on scalars, NequIP employs SE(3)-equivariant convolutions for interactions of geometric tensors, resulting in a more information-rich and faithful representation of atomic environments. The method achieves state-of-the-art accuracy on a challenging set of diverse molecules and materials while exhibiting remarkable data efficiency. NequIP outperforms existing models with up to three orders of magnitude fewer training data, challenging the widely held belief that deep neural networks require massive training sets. The high data efficiency of the method allows for the construction of accurate potentials using high-order quantum chemical level of theory as reference and enables high-fidelity molecular dynamics simulations over long time scales.
A community-powered search of machine learning strategy space to find NMR property prediction models
Aug 13, 2020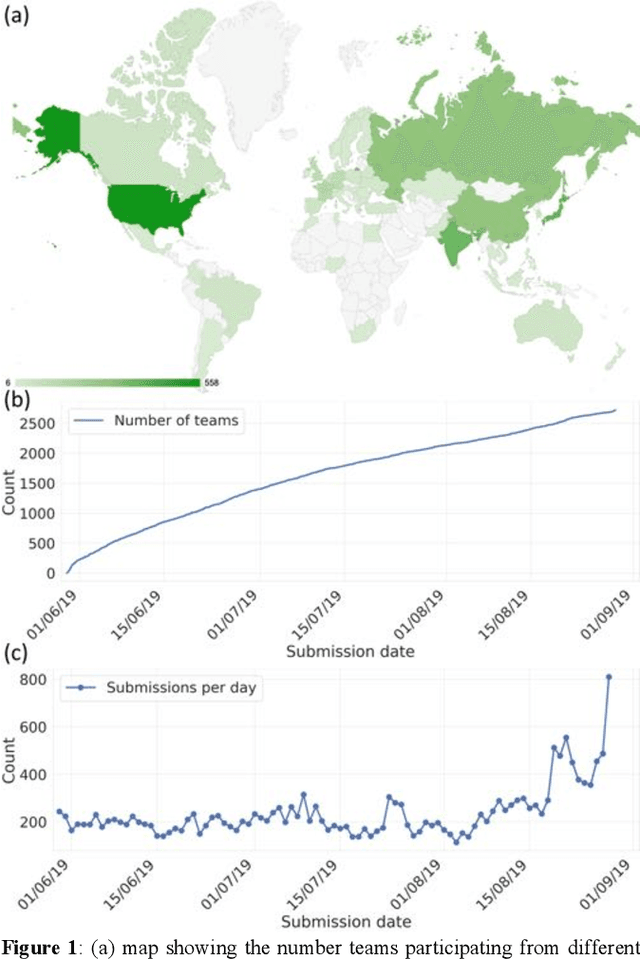
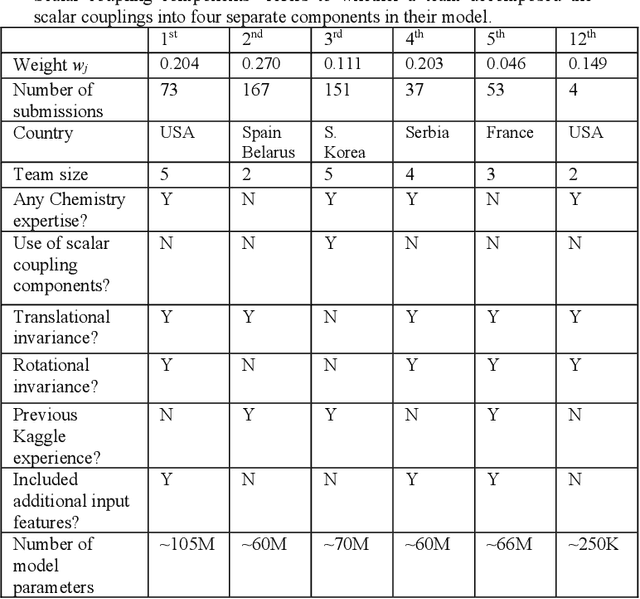
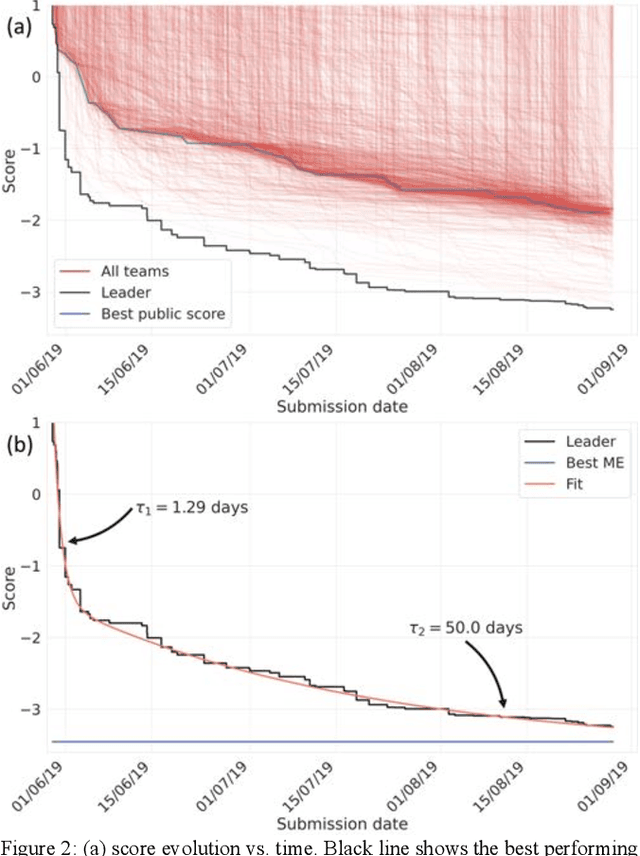
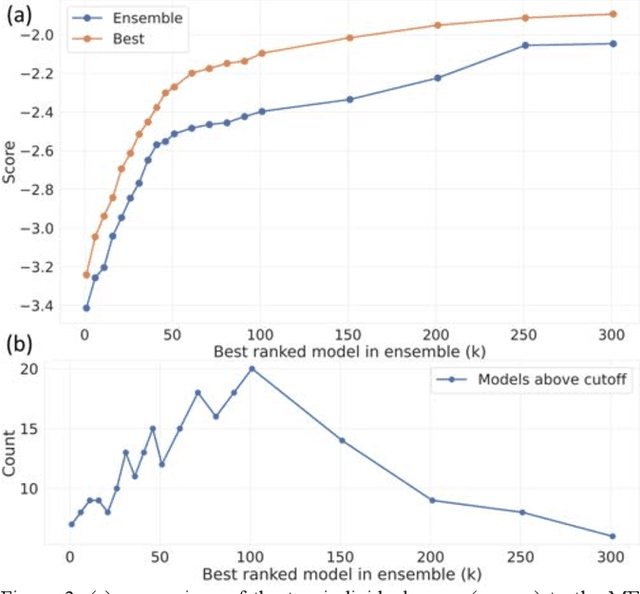
The rise of machine learning (ML) has created an explosion in the potential strategies for using data to make scientific predictions. For physical scientists wishing to apply ML strategies to a particular domain, it can be difficult to assess in advance what strategy to adopt within a vast space of possibilities. Here we outline the results of an online community-powered effort to swarm search the space of ML strategies and develop algorithms for predicting atomic-pairwise nuclear magnetic resonance (NMR) properties in molecules. Using an open-source dataset, we worked with Kaggle to design and host a 3-month competition which received 47,800 ML model predictions from 2,700 teams in 84 countries. Within 3 weeks, the Kaggle community produced models with comparable accuracy to our best previously published "in-house" efforts. A meta-ensemble model constructed as a linear combination of the top predictions has a prediction accuracy which exceeds that of any individual model, 7-19x better than our previous state-of-the-art. The results highlight the potential of transformer architectures for predicting quantum mechanical (QM) molecular properties.
Fast Neural Network Approach for Direct Covariant Forces Prediction in Complex Multi-Element Extended Systems
May 07, 2019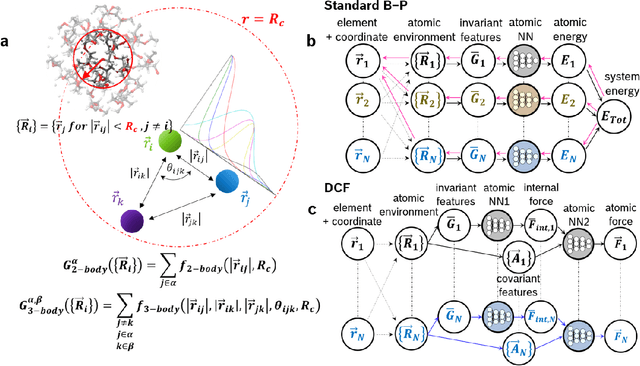
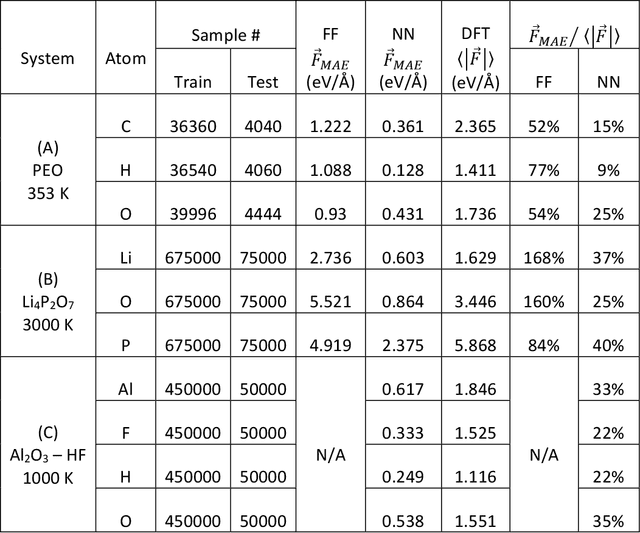
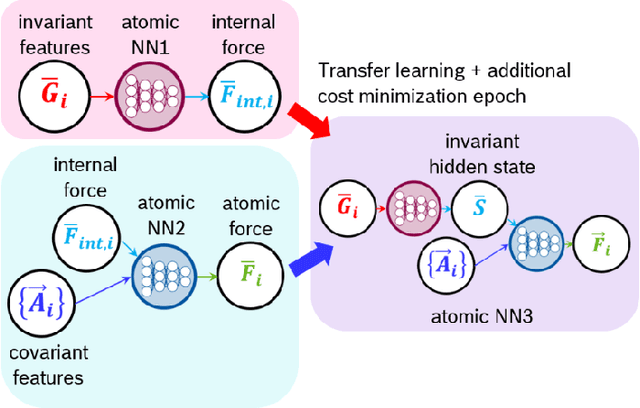
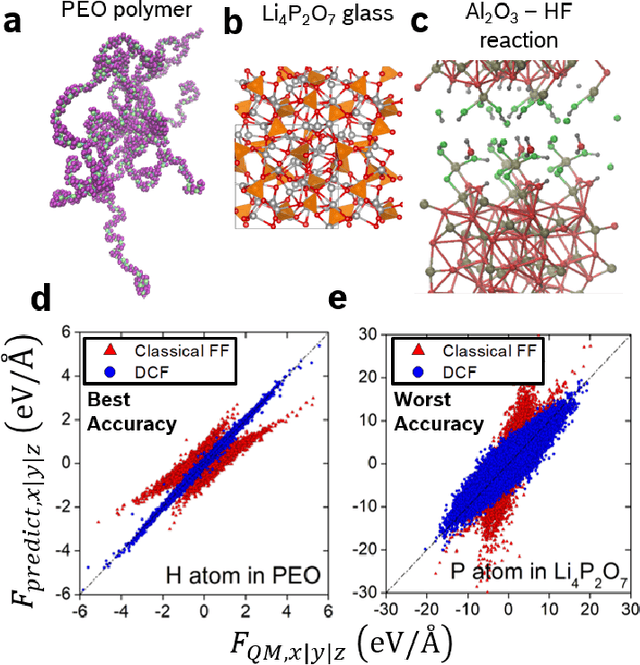
Neural network force field (NNFF) is a method for performing regression on atomic structure-force relationships, bypassing expensive quantum mechanics calculation which prevents the execution of long ab-initio quality molecular dynamics simulations. However, most NNFF methods for complex multi-element atomic systems indirectly predict atomic force vectors by exploiting just atomic structure rotation-invariant features and the network-feature spatial derivatives which are computationally expensive. We develop a staggered NNFF architecture exploiting both rotation-invariant and covariant features separately to directly predict atomic force vectors without using spatial derivatives, thereby reducing expensive structural feature calculation by ~180-480x. This acceleration enables us to develop NNFF which directly predicts atomic forces in complex ternary and quaternary-element extended systems comprised of long polymer chains, amorphous oxide, and surface chemical reactions. The staggered rotation-invariant-covariant architecture described here can also directly predict complex covariant vector outputs from local physical structures in domains beyond computational material science.