 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards Symbolic XAI -- Explanation Through Human Understandable Logical Relationships Between Features
Aug 30, 2024Explainable Artificial Intelligence (XAI) plays a crucial role in fostering transparency and trust in AI systems, where traditional XAI approaches typically offer one level of abstraction for explanations, often in the form of heatmaps highlighting single or multiple input features. However, we ask whether abstract reasoning or problem-solving strategies of a model may also be relevant, as these align more closely with how humans approach solutions to problems. We propose a framework, called Symbolic XAI, that attributes relevance to symbolic queries expressing logical relationships between input features, thereby capturing the abstract reasoning behind a model's predictions. The methodology is built upon a simple yet general multi-order decomposition of model predictions. This decomposition can be specified using higher-order propagation-based relevance methods, such as GNN-LRP, or perturbation-based explanation methods commonly used in XAI. The effectiveness of our framework is demonstrated in the domains of natural language processing (NLP), vision, and quantum chemistry (QC), where abstract symbolic domain knowledge is abundant and of significant interest to users. The Symbolic XAI framework provides an understanding of the model's decision-making process that is both flexible for customization by the user and human-readable through logical formulas.
SchNetPack 2.0: A neural network toolbox for atomistic machine learning
Dec 11, 2022



SchNetPack is a versatile neural networks toolbox that addresses both the requirements of method development and application of atomistic machine learning. Version 2.0 comes with an improved data pipeline, modules for equivariant neural networks as well as a PyTorch implementation of molecular dynamics. An optional integration with PyTorch Lightning and the Hydra configuration framework powers a flexible command-line interface. This makes SchNetPack 2.0 easily extendable with custom code and ready for complex training task such as generation of 3d molecular structures.
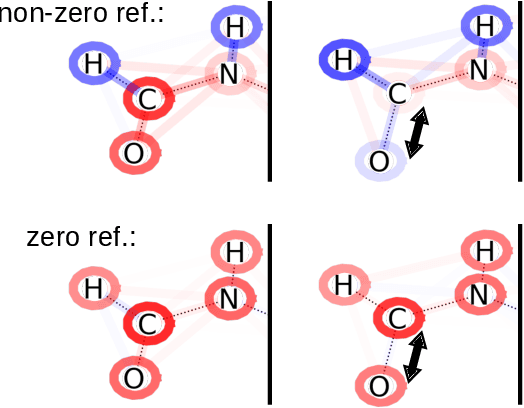
Automatic Identification of Chemical Moieties
Mar 30, 2022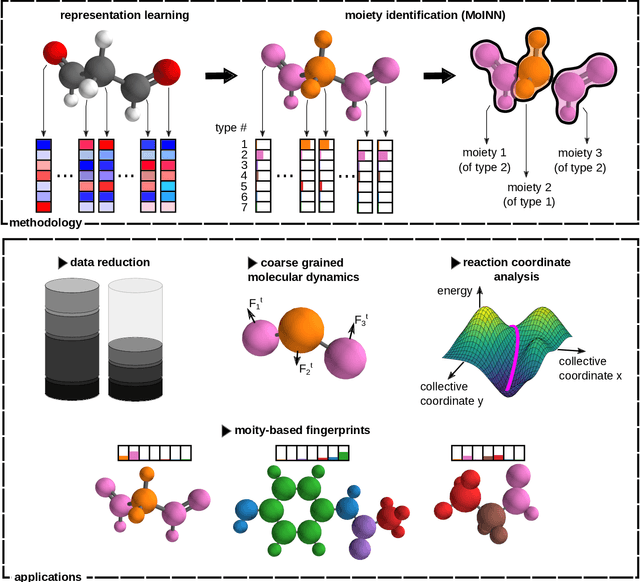
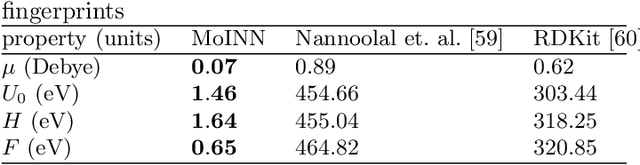
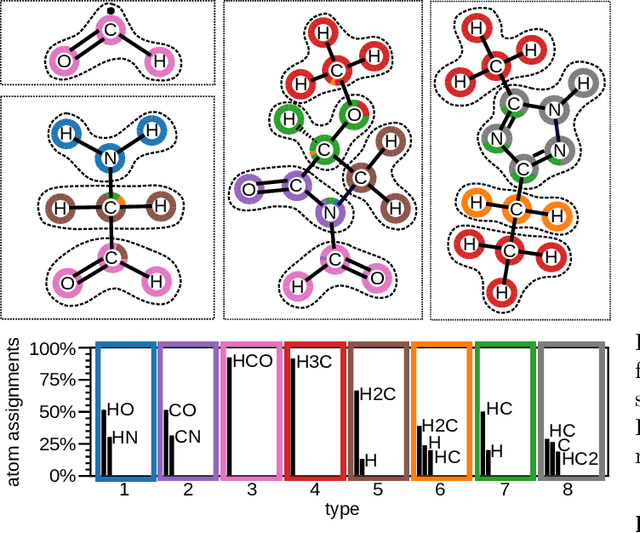
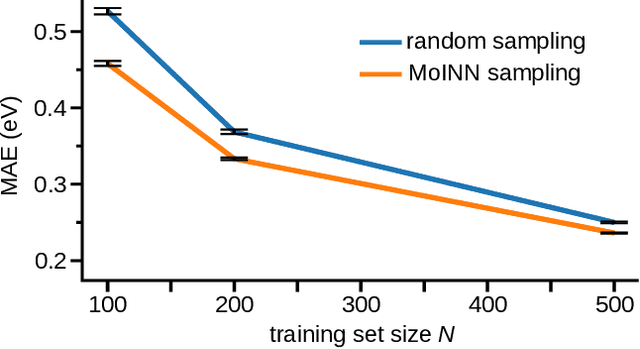
In recent years, the prediction of quantum mechanical observables with machine learning methods has become increasingly popular. Message-passing neural networks (MPNNs) solve this task by constructing atomic representations, from which the properties of interest are predicted. Here, we introduce a method to automatically identify chemical moieties (molecular building blocks) from such representations, enabling a variety of applications beyond property prediction, which otherwise rely on expert knowledge. The required representation can either be provided by a pretrained MPNN, or learned from scratch using only structural information. Beyond the data-driven design of molecular fingerprints, the versatility of our approach is demonstrated by enabling the selection of representative entries in chemical databases, the automatic construction of coarse-grained force fields, as well as the identification of reaction coordinates.
Toward Explainable AI for Regression Models
Dec 21, 2021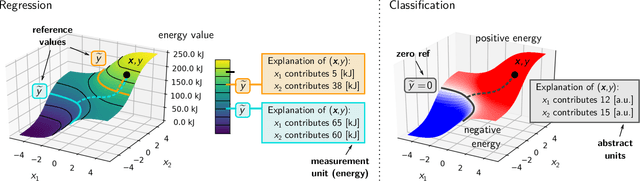
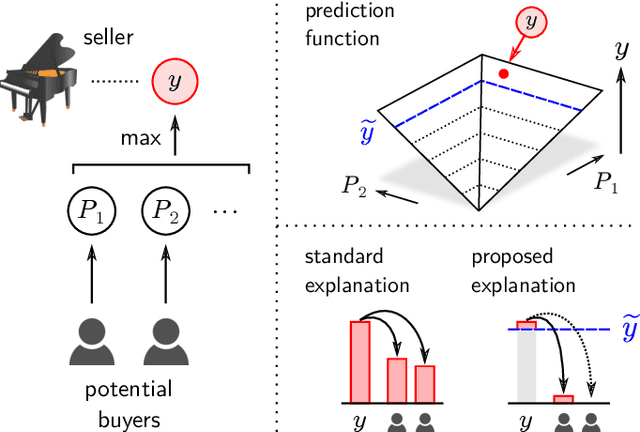
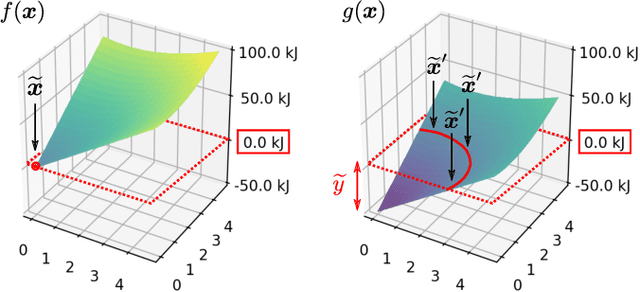
In addition to the impressive predictive power of machine learning (ML) models, more recently, explanation methods have emerged that enable an interpretation of complex non-linear learning models such as deep neural networks. Gaining a better understanding is especially important e.g. for safety-critical ML applications or medical diagnostics etc. While such Explainable AI (XAI) techniques have reached significant popularity for classifiers, so far little attention has been devoted to XAI for regression models (XAIR). In this review, we clarify the fundamental conceptual differences of XAI for regression and classification tasks, establish novel theoretical insights and analysis for XAIR, provide demonstrations of XAIR on genuine practical regression problems, and finally discuss the challenges remaining for the field.
XAI for Graphs: Explaining Graph Neural Network Predictions by Identifying Relevant Walks
Jun 12, 2020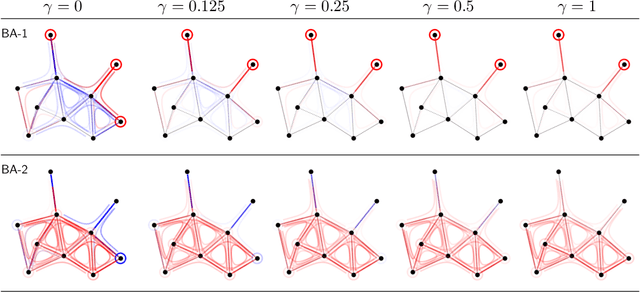
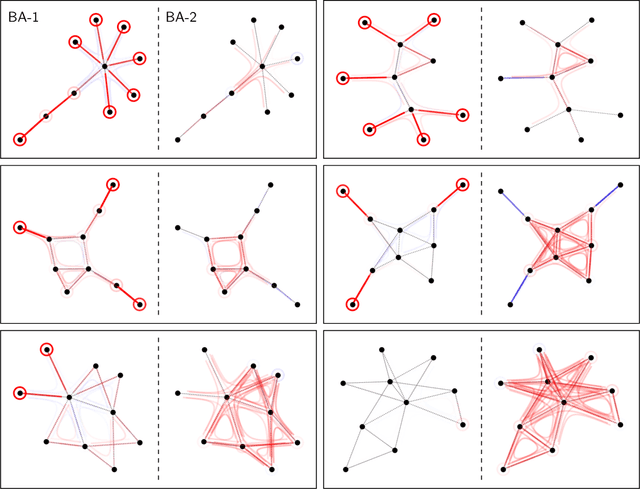
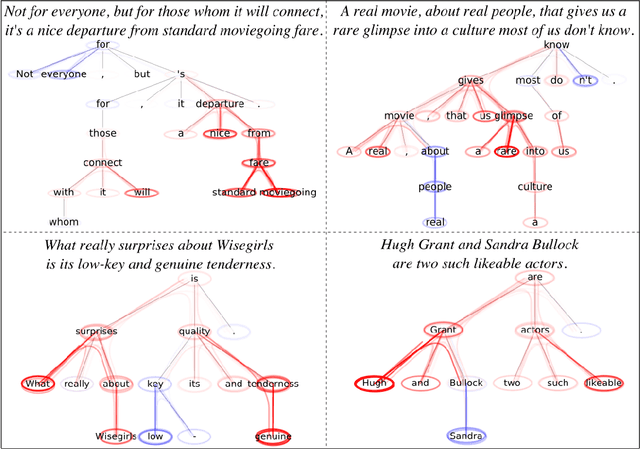
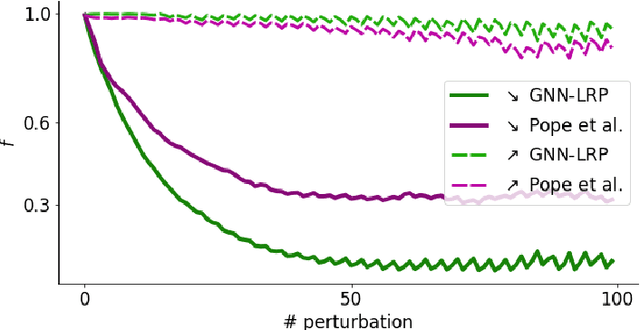
Graph Neural Networks (GNNs) are a popular approach for predicting graph structured data. As GNNs tightly entangle the input graph into the neural network structure, common explainable AI (XAI) approaches are not applicable. To a large extent, GNNs have remained black-boxes for the user so far. In this paper, we contribute by proposing a new XAI approach for GNNs. Our approach is derived from high-order Taylor expansions and is able to generate a decomposition of the GNN prediction as a collection of relevant walks on the input graph. We find that these high-order Taylor expansions can be equivalently (and more simply) computed using multiple backpropagation passes from the top layer of the GNN to the first layer. The explanation can then be further robustified and generalized by using layer-wise-relevance propagation (LRP) in place of the standard equations for gradient propagation. Our novel method which we denote as 'GNN-LRP' is tested on scale-free graphs, sentence parsing trees, molecular graphs, and pixel lattices representing images. In each case, it performs stably and accurately, and delivers interesting and novel application insights.