 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRNA-FM: Flow-Matching Generative Model for Genome-wide RNA-Seq Prediction
May 12, 2026Histopathology whole-slide images (WSIs) are routinely acquired in clinical practice and contain rich tissue morphology but lack direct molecular architecture and functional programs defining pathological states, whereas RNA sequencing (RNA-seq) provides genome-wide transcriptional profiles at substantial cost, thereby motivating WSI-based genome-wide transcriptomic prediction. Existing approaches for predicting gene expression from WSIs predominantly rely on deterministic regression with one-to-one mapping, limiting their ability to capture biological heterogeneity and predictive uncertainty. We propose RNA-FM, a flow-matching generative framework for genome-wide bulk RNA-seq prediction from WSIs. RNA-FM formulates transcriptomic prediction as a continuous-time conditional transport problem, learning a velocity field that maps a simple prior to the target gene expression distribution conditioned on morphologies. By integrating pathway-level structure, RNA-FM enables scalable and biologically interpretable genome-wide gene expression imputation. Extensive experiments demonstrate that RNA-FM consistently outperforms state-of-the-art approaches while maintaining biological meaningfulness. Code is available at https://github.com/YXSong000/RNA-FM.
GuiDINO: Rethinking Vision Foundation Model in Medical Image Segmentation
Mar 01, 2026Foundation vision models are increasingly adopted in medical image analysis. Due to domain shift, these pretrained models misalign with medical image segmentation needs without being fully fine-tuned or lightly adapted. We introduce GuiDINO, a framework that repositions native foundation model to acting as a visual guidance generator for downstream segmentation. GuiDINO extracts visual feature representation from DINOv3 and converts them into a spatial guide mask via a lightweight TokenBook mechanism, which aggregates token-prototype similarities. This guide mask gates feature activations in multiple segmentation backbones, thereby injecting foundation-model priors while preserving the inductive biases and efficiency of medical dedicated architectures. Training relies on a guide supervision objective loss that aligns the guide mask to ground-truth regions, optionally augmented by a boundary-focused hinge loss to sharpen fine structures. GuiDINO also supports parameter-efficient adaptation through LoRA on the DINOv3 guide backbone. Across diverse medical datasets and nnUNet-style inference, GuiDINO consistently improves segmentation quality and boundary robustness, suggesting a practical alternative to fine-tuning and offering a new perspective on how foundation models can best serve medical vision. Code is available at https://github.com/Hi-FishU/GuiDINO
ScSAM: Debiasing Morphology and Distributional Variability in Subcellular Semantic Segmentation
Jul 23, 2025The significant morphological and distributional variability among subcellular components poses a long-standing challenge for learning-based organelle segmentation models, significantly increasing the risk of biased feature learning. Existing methods often rely on single mapping relationships, overlooking feature diversity and thereby inducing biased training. Although the Segment Anything Model (SAM) provides rich feature representations, its application to subcellular scenarios is hindered by two key challenges: (1) The variability in subcellular morphology and distribution creates gaps in the label space, leading the model to learn spurious or biased features. (2) SAM focuses on global contextual understanding and often ignores fine-grained spatial details, making it challenging to capture subtle structural alterations and cope with skewed data distributions. To address these challenges, we introduce ScSAM, a method that enhances feature robustness by fusing pre-trained SAM with Masked Autoencoder (MAE)-guided cellular prior knowledge to alleviate training bias from data imbalance. Specifically, we design a feature alignment and fusion module to align pre-trained embeddings to the same feature space and efficiently combine different representations. Moreover, we present a cosine similarity matrix-based class prompt encoder to activate class-specific features to recognize subcellular categories. Extensive experiments on diverse subcellular image datasets demonstrate that ScSAM outperforms state-of-the-art methods.
Revisiting Adaptive Cellular Recognition Under Domain Shifts: A Contextual Correspondence View
Jul 19, 2024



Cellular nuclei recognition serves as a fundamental and essential step in the workflow of digital pathology. However, with disparate source organs and staining procedures among histology image clusters, the scanned tiles inherently conform to a non-uniform data distribution, which induces deteriorated promises for general cross-cohort usages. Despite the latest efforts leveraging domain adaptation to mitigate distributional discrepancy, those methods are subjected to modeling the morphological characteristics of each cell individually, disregarding the hierarchical latent structure and intrinsic contextual correspondences across the tumor micro-environment. In this work, we identify the importance of implicit correspondences across biological contexts for exploiting domain-invariant pathological composition and thereby propose to exploit the dependence over various biological structures for domain adaptive cellular recognition. We discover those high-level correspondences via unsupervised contextual modeling and use them as bridges to facilitate adaptation over diverse organs and stains. In addition, to further exploit the rich spatial contexts embedded amongst nuclear communities, we propose self-adaptive dynamic distillation to secure instance-aware trade-offs across different model constituents. The proposed method is extensively evaluated on a broad spectrum of cross-domain settings under miscellaneous data distribution shifts and outperforms the state-of-the-art methods by a substantial margin. Code is available at https://github.com/camwew/CellularRecognition_DA_CC.
Seeing Unseen: Discover Novel Biomedical Concepts via Geometry-Constrained Probabilistic Modeling
Mar 05, 2024



Machine learning holds tremendous promise for transforming the fundamental practice of scientific discovery by virtue of its data-driven nature. With the ever-increasing stream of research data collection, it would be appealing to autonomously explore patterns and insights from observational data for discovering novel classes of phenotypes and concepts. However, in the biomedical domain, there are several challenges inherently presented in the cumulated data which hamper the progress of novel class discovery. The non-i.i.d. data distribution accompanied by the severe imbalance among different groups of classes essentially leads to ambiguous and biased semantic representations. In this work, we present a geometry-constrained probabilistic modeling treatment to resolve the identified issues. First, we propose to parameterize the approximated posterior of instance embedding as a marginal von MisesFisher distribution to account for the interference of distributional latent bias. Then, we incorporate a suite of critical geometric properties to impose proper constraints on the layout of constructed embedding space, which in turn minimizes the uncontrollable risk for unknown class learning and structuring. Furthermore, a spectral graph-theoretic method is devised to estimate the number of potential novel classes. It inherits two intriguing merits compared to existent approaches, namely high computational efficiency and flexibility for taxonomy-adaptive estimation. Extensive experiments across various biomedical scenarios substantiate the effectiveness and general applicability of our method.
Learning to Generalize over Subpartitions for Heterogeneity-aware Domain Adaptive Nuclei Segmentation
Jan 21, 2024



Annotation scarcity and cross-modality/stain data distribution shifts are two major obstacles hindering the application of deep learning models for nuclei analysis, which holds a broad spectrum of potential applications in digital pathology. Recently, unsupervised domain adaptation (UDA) methods have been proposed to mitigate the distributional gap between different imaging modalities for unsupervised nuclei segmentation in histopathology images. However, existing UDA methods are built upon the assumption that data distributions within each domain should be uniform. Based on the over-simplified supposition, they propose to align the histopathology target domain with the source domain integrally, neglecting severe intra-domain discrepancy over subpartitions incurred by mixed cancer types and sampling organs. In this paper, for the first time, we propose to explicitly consider the heterogeneity within the histopathology domain and introduce open compound domain adaptation (OCDA) to resolve the crux. In specific, a two-stage disentanglement framework is proposed to acquire domain-invariant feature representations at both image and instance levels. The holistic design addresses the limitations of existing OCDA approaches which struggle to capture instance-wise variations. Two regularization strategies are specifically devised herein to leverage the rich subpartition-specific characteristics in histopathology images and facilitate subdomain decomposition. Moreover, we propose a dual-branch nucleus shape and structure preserving module to prevent nucleus over-generation and deformation in the synthesized images. Experimental results on both cross-modality and cross-stain scenarios over a broad range of diverse datasets demonstrate the superiority of our method compared with state-of-the-art UDA and OCDA methods.
Taxonomy Adaptive Cross-Domain Adaptation in Medical Imaging via Optimization Trajectory Distillation
Jul 27, 2023



The success of automated medical image analysis depends on large-scale and expert-annotated training sets. Unsupervised domain adaptation (UDA) has been raised as a promising approach to alleviate the burden of labeled data collection. However, they generally operate under the closed-set adaptation setting assuming an identical label set between the source and target domains, which is over-restrictive in clinical practice where new classes commonly exist across datasets due to taxonomic inconsistency. While several methods have been presented to tackle both domain shifts and incoherent label sets, none of them take into account the common characteristics of the two issues and consider the learning dynamics along network training. In this work, we propose optimization trajectory distillation, a unified approach to address the two technical challenges from a new perspective. It exploits the low-rank nature of gradient space and devises a dual-stream distillation algorithm to regularize the learning dynamics of insufficiently annotated domain and classes with the external guidance obtained from reliable sources. Our approach resolves the issue of inadequate navigation along network optimization, which is the major obstacle in the taxonomy adaptive cross-domain adaptation scenario. We evaluate the proposed method extensively on several tasks towards various endpoints with clinical and open-world significance. The results demonstrate its effectiveness and improvements over previous methods.
NaroNet: Discovery of tumor microenvironment elements from highly multiplexed images
Mar 25, 2021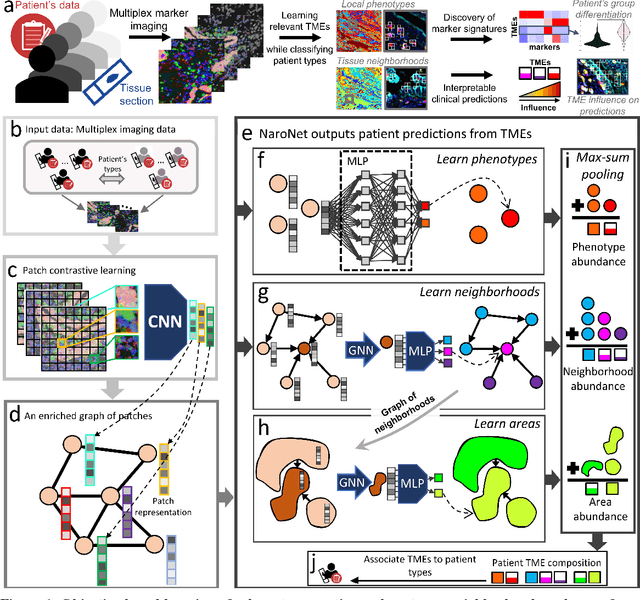
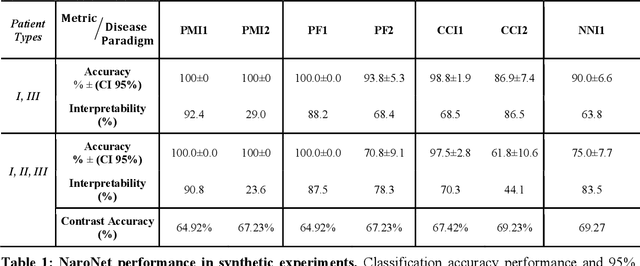
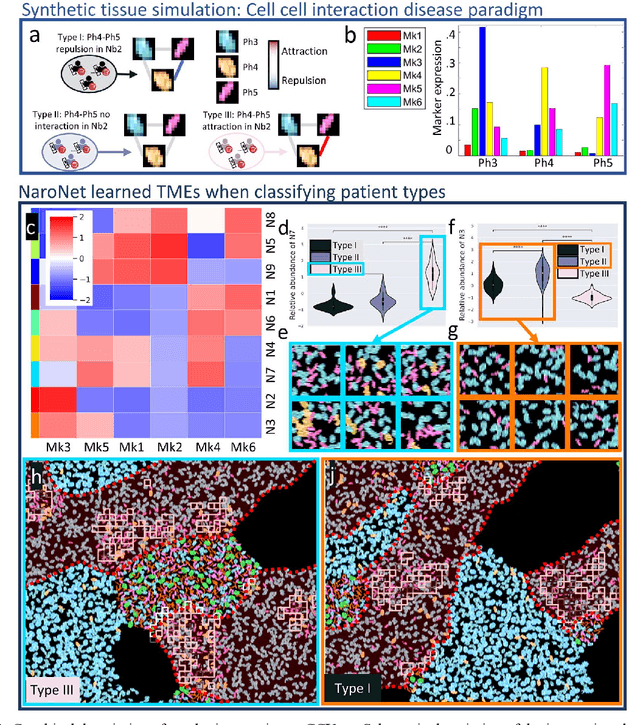
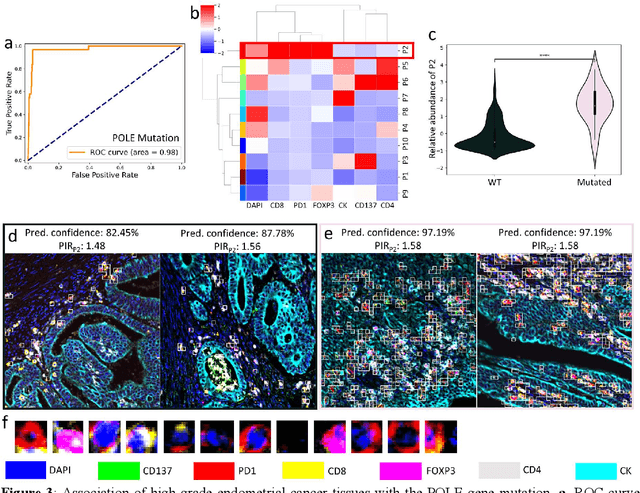
Many efforts have been made to discover tumor-specific microenvironment elements (TMEs) from immunostained tissue sections. However, the identification of yet unknown but relevant TMEs from multiplex immunostained tissues remains a challenge, due to the number of markers involved (tens) and the complexity of their spatial interactions. We present NaroNet, which uses machine learning to identify and annotate known as well as novel TMEs from self-supervised embeddings of cells, organized at different levels (local cell phenotypes and cellular neighborhoods). Then it uses the abundance of TMEs to classify patients based on biological or clinical features. We validate NaroNet using synthetic patient cohorts with adjustable incidence of different TMEs and two cancer patient datasets. In both synthetic and real datasets, NaroNet unsupervisedly identifies novel TMEs, relevant for the user-defined classification task. As NaroNet requires only patient-level information, it renders state-of-the-art computational methods accessible to a broad audience, accelerating the discovery of biomarker signatures.