 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEfficient Low-Order Approximation of First-Passage Time Distributions
Nov 01, 2017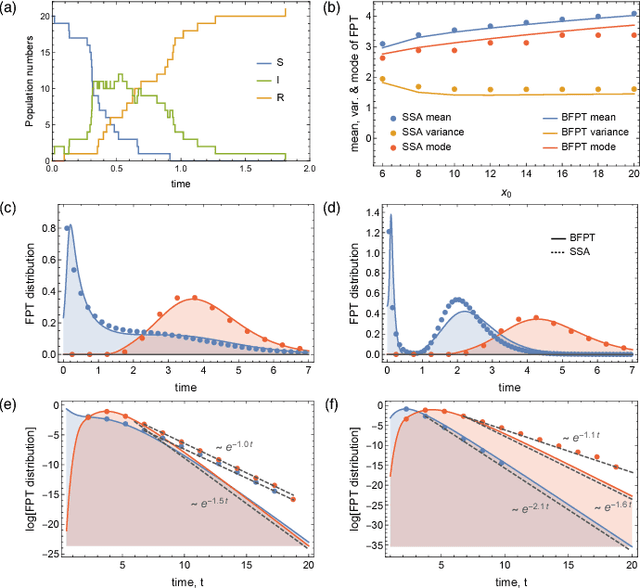
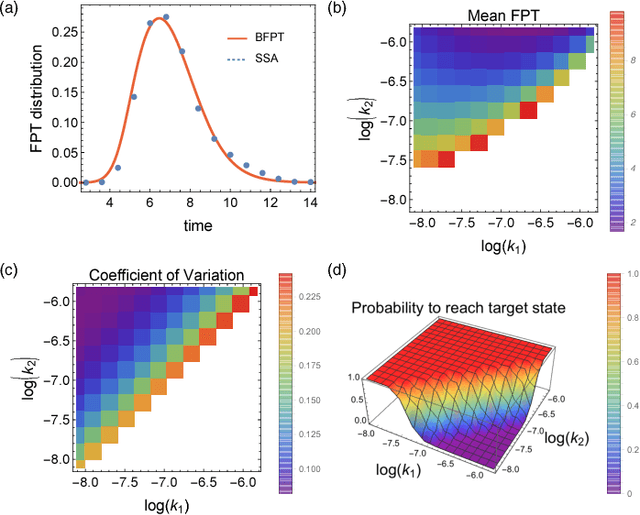
We consider the problem of computing first-passage time distributions for reaction processes modelled by master equations. We show that this generally intractable class of problems is equivalent to a sequential Bayesian inference problem for an auxiliary observation process. The solution can be approximated efficiently by solving a closed set of coupled ordinary differential equations (for the low-order moments of the process) whose size scales with the number of species. We apply it to an epidemic model and a trimerisation process, and show good agreement with stochastic simulations.
* 5 pages, 3 figures
On learning the structure of Bayesian Networks and submodular function maximization
Jun 07, 2017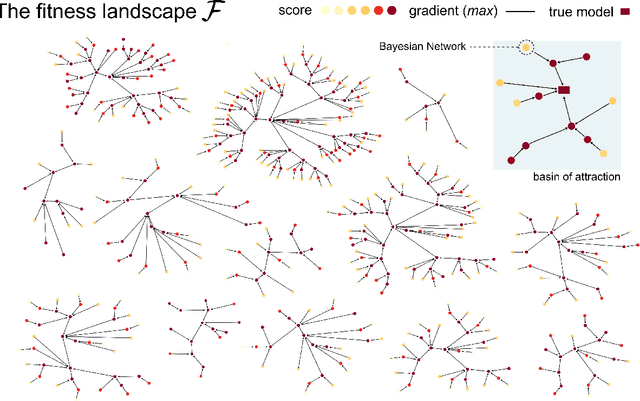
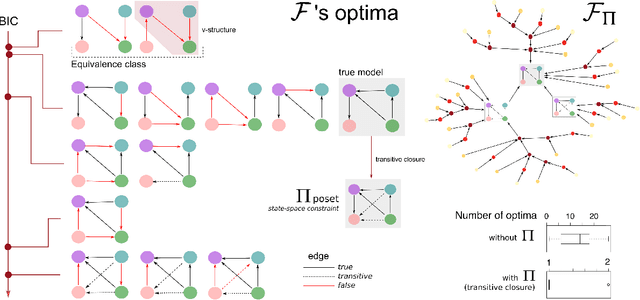
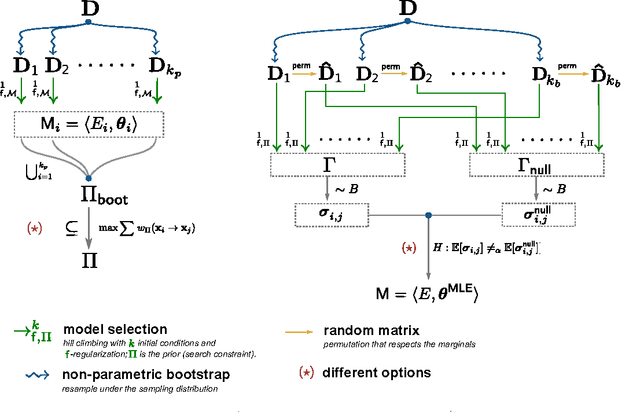
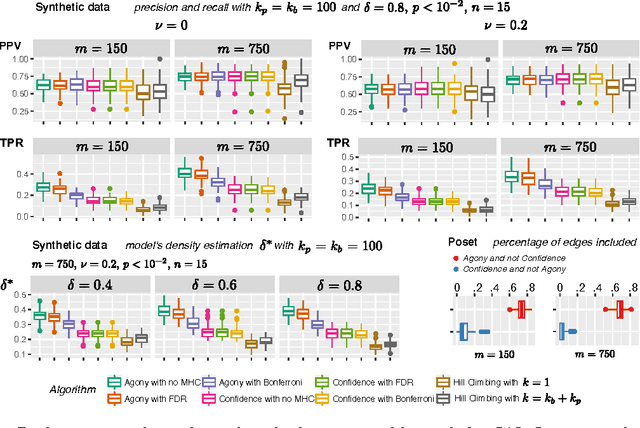
Learning the structure of dependencies among multiple random variables is a problem of considerable theoretical and practical interest. In practice, score optimisation with multiple restarts provides a practical and surprisingly successful solution, yet the conditions under which this may be a well founded strategy are poorly understood. In this paper, we prove that the problem of identifying the structure of a Bayesian Network via regularised score optimisation can be recast, in expectation, as a submodular optimisation problem, thus guaranteeing optimality with high probability. This result both explains the practical success of optimisation heuristics, and suggests a way to improve on such algorithms by artificially simulating multiple data sets via a bootstrap procedure. We show on several synthetic data sets that the resulting algorithm yields better recovery performance than the state of the art, and illustrate in a real cancer genomic study how such an approach can lead to valuable practical insights.
Approximation and inference methods for stochastic biochemical kinetics - a tutorial review
Jan 12, 2017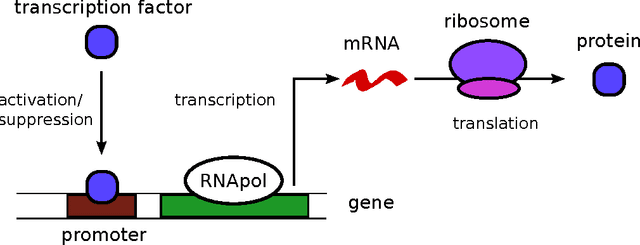
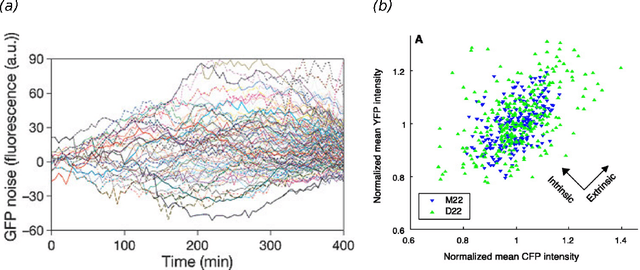
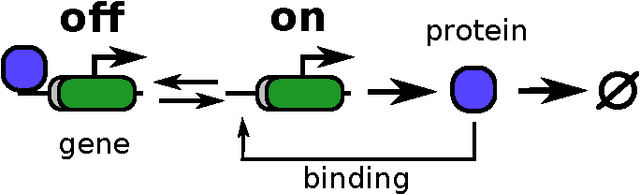
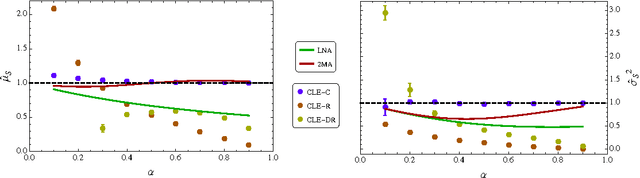
Stochastic fluctuations of molecule numbers are ubiquitous in biological systems. Important examples include gene expression and enzymatic processes in living cells. Such systems are typically modelled as chemical reaction networks whose dynamics are governed by the Chemical Master Equation. Despite its simple structure, no analytic solutions to the Chemical Master Equation are known for most systems. Moreover, stochastic simulations are computationally expensive, making systematic analysis and statistical inference a challenging task. Consequently, significant effort has been spent in recent decades on the development of efficient approximation and inference methods. This article gives an introduction to basic modelling concepts as well as an overview of state of the art methods. First, we motivate and introduce deterministic and stochastic methods for modelling chemical networks, and give an overview of simulation and exact solution methods. Next, we discuss several approximation methods, including the chemical Langevin equation, the system size expansion, moment closure approximations, time-scale separation approximations and hybrid methods. We discuss their various properties and review recent advances and remaining challenges for these methods. We present a comparison of several of these methods by means of a numerical case study and highlight some of their respective advantages and disadvantages. Finally, we discuss the problem of inference from experimental data in the Bayesian framework and review recent methods developed the literature. In summary, this review gives a self-contained introduction to modelling, approximations and inference methods for stochastic chemical kinetics.
Property-driven State-Space Coarsening for Continuous Time Markov Chains
Oct 29, 2016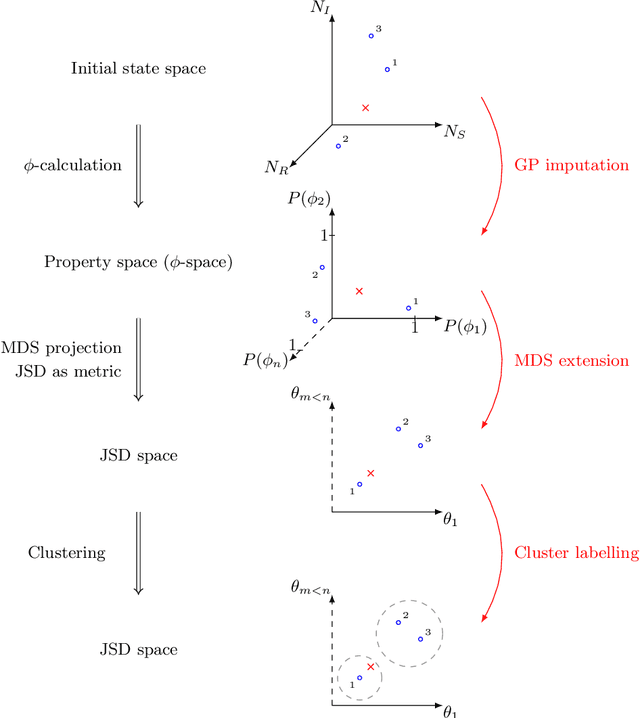
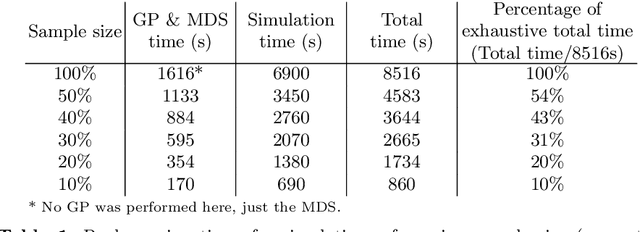
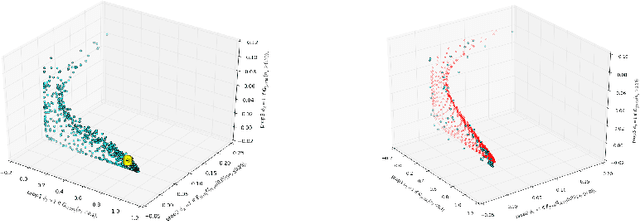
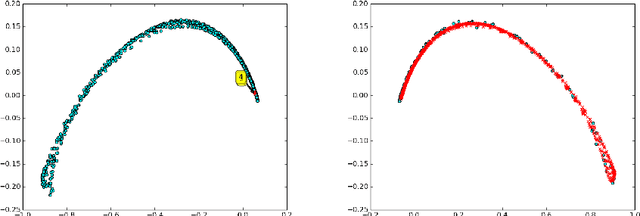
Dynamical systems with large state-spaces are often expensive to thoroughly explore experimentally. Coarse-graining methods aim to define simpler systems which are more amenable to analysis and exploration; most current methods, however, focus on a priori state aggregation based on similarities in transition rates, which is not necessarily reflected in similar behaviours at the level of trajectories. We propose a way to coarsen the state-space of a system which optimally preserves the satisfaction of a set of logical specifications about the system's trajectories. Our approach is based on Gaussian Process emulation and Multi-Dimensional Scaling, a dimensionality reduction technique which optimally preserves distances in non-Euclidean spaces. We show how to obtain low-dimensional visualisations of the system's state-space from the perspective of properties' satisfaction, and how to define macro-states which behave coherently with respect to the specifications. Our approach is illustrated on a non-trivial running example, showing promising performance and high computational efficiency.
* 16 pages, 6 figures, 1 table
Cox process representation and inference for stochastic reaction-diffusion processes
Aug 22, 2016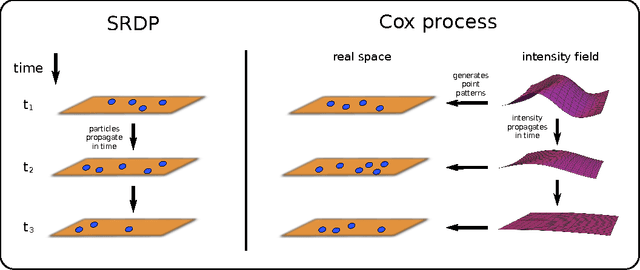
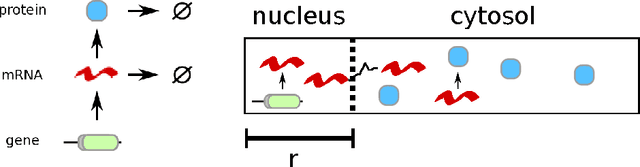
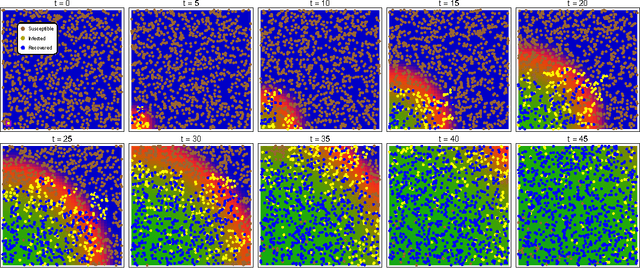
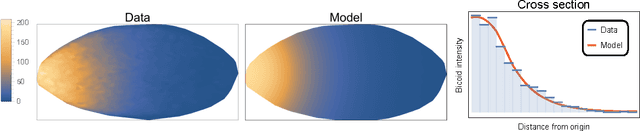
Complex behaviour in many systems arises from the stochastic interactions of spatially distributed particles or agents. Stochastic reaction-diffusion processes are widely used to model such behaviour in disciplines ranging from biology to the social sciences, yet they are notoriously difficult to simulate and calibrate to observational data. Here we use ideas from statistical physics and machine learning to provide a solution to the inverse problem of learning a stochastic reaction-diffusion process from data. Our solution relies on a non-trivial connection between stochastic reaction-diffusion processes and spatio-temporal Cox processes, a well-studied class of models from computational statistics. This connection leads to an efficient and flexible algorithm for parameter inference and model selection. Our approach shows excellent accuracy on numeric and real data examples from systems biology and epidemiology. Our work provides both insights into spatio-temporal stochastic systems, and a practical solution to a long-standing problem in computational modelling.
* 18 pages, 5 figures
Expectation propagation for continuous time stochastic processes
Jun 28, 2016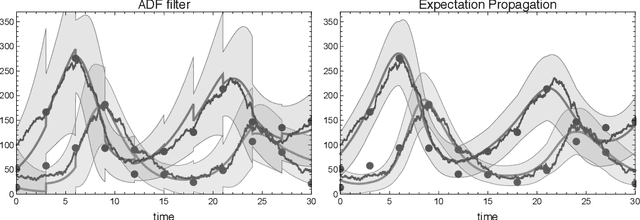
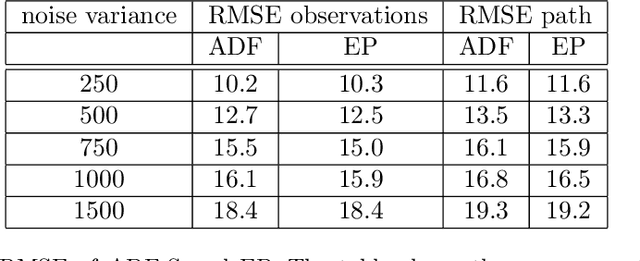
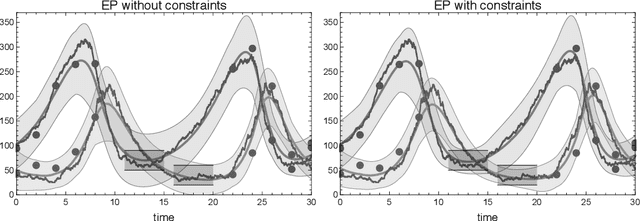
We consider the inverse problem of reconstructing the posterior measure over the trajec- tories of a diffusion process from discrete time observations and continuous time constraints. We cast the problem in a Bayesian framework and derive approximations to the posterior distributions of single time marginals using variational approximate inference. We then show how the approximation can be extended to a wide class of discrete-state Markov jump pro- cesses by making use of the chemical Langevin equation. Our empirical results show that the proposed method is computationally efficient and provides good approximations for these classes of inverse problems.
Unbiased Bayesian Inference for Population Markov Jump Processes via Random Truncations
May 13, 2016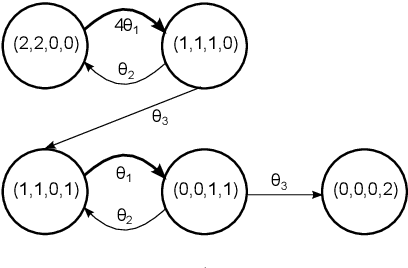

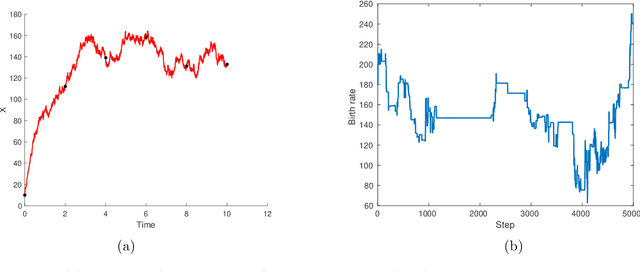
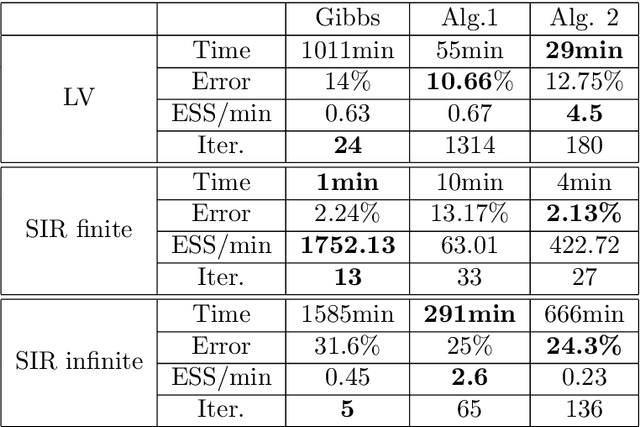
We consider continuous time Markovian processes where populations of individual agents interact stochastically according to kinetic rules. Despite the increasing prominence of such models in fields ranging from biology to smart cities, Bayesian inference for such systems remains challenging, as these are continuous time, discrete state systems with potentially infinite state-space. Here we propose a novel efficient algorithm for joint state / parameter posterior sampling in population Markov Jump processes. We introduce a class of pseudo-marginal sampling algorithms based on a random truncation method which enables a principled treatment of infinite state spaces. Extensive evaluation on a number of benchmark models shows that this approach achieves considerable savings compared to state of the art methods, retaining accuracy and fast convergence. We also present results on a synthetic biology data set showing the potential for practical usefulness of our work.
Matching models across abstraction levels with Gaussian Processes
May 07, 2016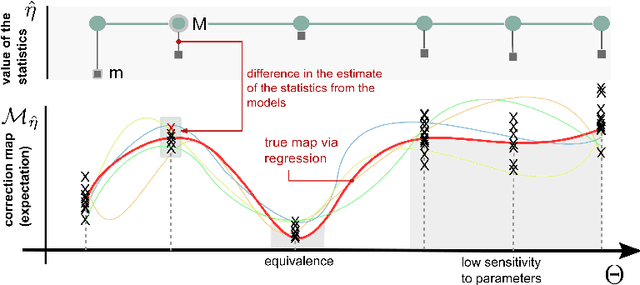
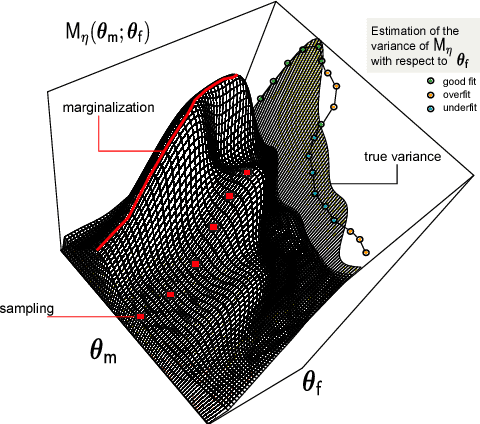
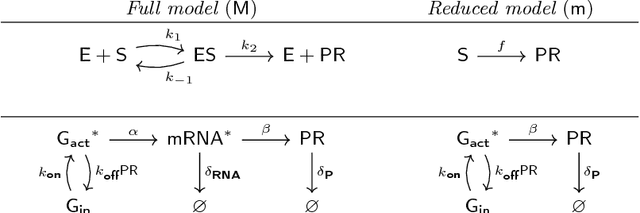
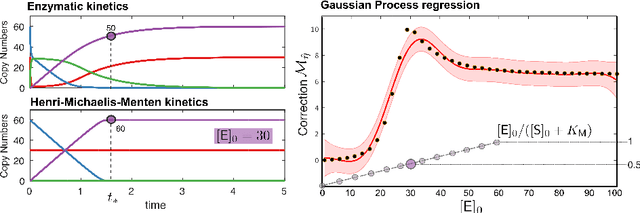
Biological systems are often modelled at different levels of abstraction depending on the particular aims/resources of a study. Such different models often provide qualitatively concordant predictions over specific parametrisations, but it is generally unclear whether model predictions are quantitatively in agreement, and whether such agreement holds for different parametrisations. Here we present a generally applicable statistical machine learning methodology to automatically reconcile the predictions of different models across abstraction levels. Our approach is based on defining a correction map, a random function which modifies the output of a model in order to match the statistics of the output of a different model of the same system. We use two biological examples to give a proof-of-principle demonstration of the methodology, and discuss its advantages and potential further applications.
Sparse Approximate Inference for Spatio-Temporal Point Process Models
Jul 06, 2015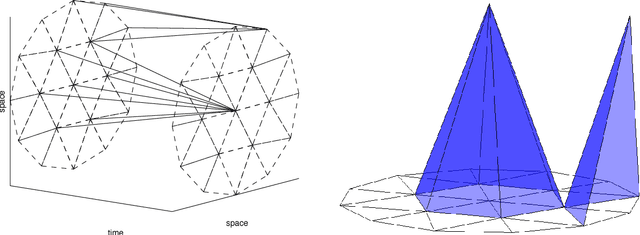
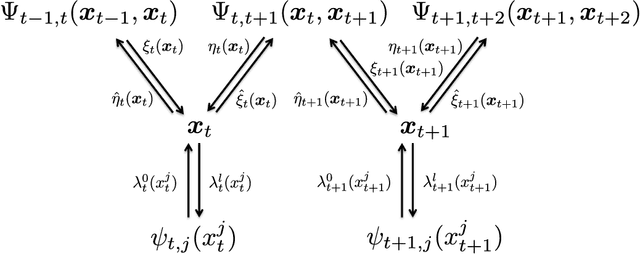
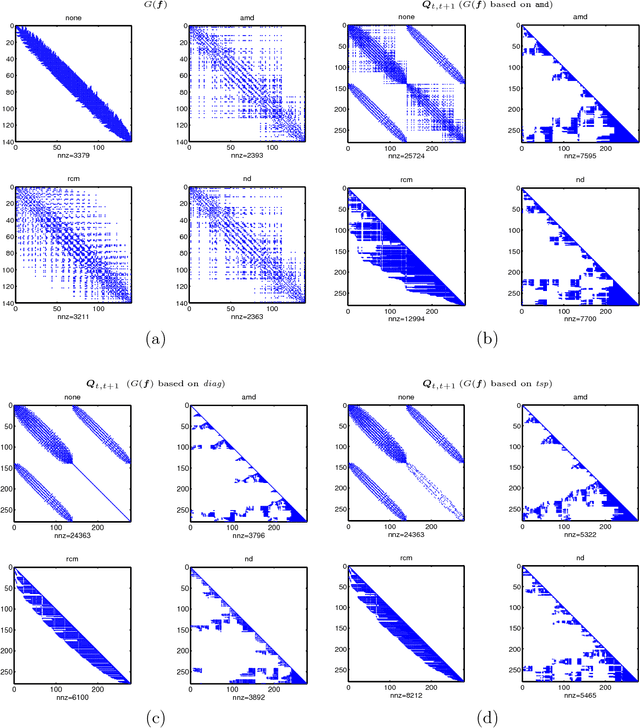
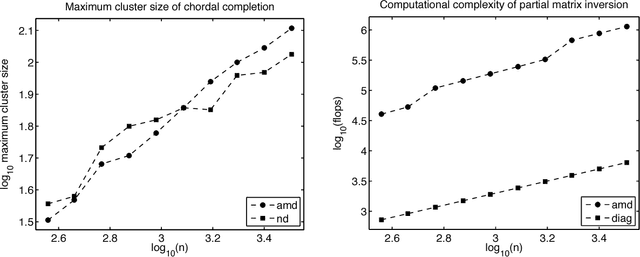
Spatio-temporal point process models play a central role in the analysis of spatially distributed systems in several disciplines. Yet, scalable inference remains computa- tionally challenging both due to the high resolution modelling generally required and the analytically intractable likelihood function. Here, we exploit the sparsity structure typical of (spatially) discretised log-Gaussian Cox process models by using approximate message-passing algorithms. The proposed algorithms scale well with the state dimension and the length of the temporal horizon with moderate loss in distributional accuracy. They hence provide a flexible and faster alternative to both non-linear filtering-smoothing type algorithms and to approaches that implement the Laplace method or expectation propagation on (block) sparse latent Gaussian models. We infer the parameters of the latent Gaussian model using a structured variational Bayes approach. We demonstrate the proposed framework on simulation studies with both Gaussian and point-process observations and use it to reconstruct the conflict intensity and dynamics in Afghanistan from the WikiLeaks Afghan War Diary.
Learning Temporal Logical Properties Discriminating ECG models of Cardiac Arrhytmias
Dec 29, 2013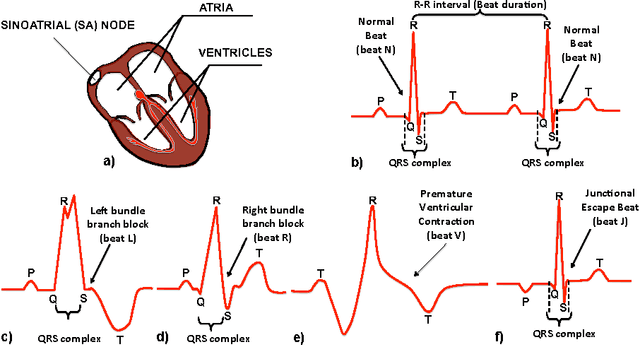

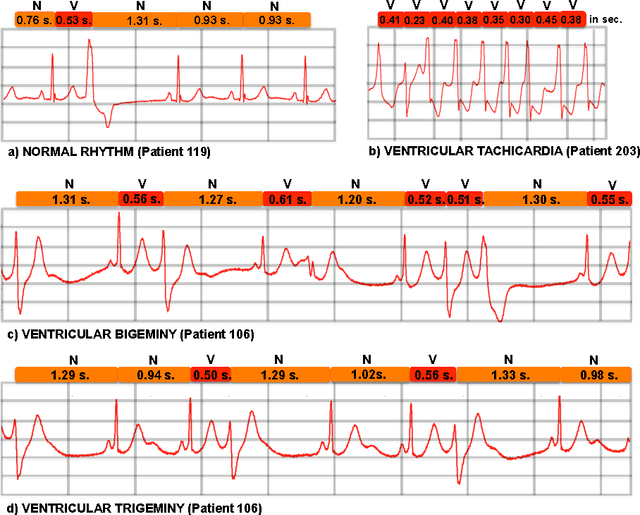
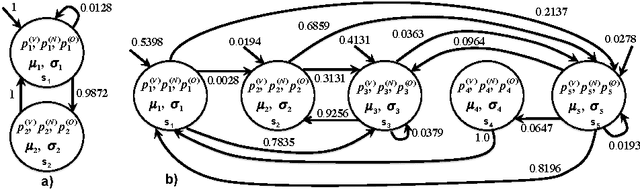
We present a novel approach to learn the formulae characterising the emergent behaviour of a dynamical system from system observations. At a high level, the approach starts by devising a statistical dynamical model of the system which optimally fits the observations. We then propose general optimisation strategies for selecting high support formulae (under the learnt model of the system) either within a discrete set of formulae of bounded complexity, or a parametric family of formulae. We illustrate and apply the methodology on an in-depth case study of characterising cardiac malfunction from electro-cardiogram data, where our approach enables us to quantitatively determine the diagnostic power of a formula in discriminating between different cardiac conditions.