 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Diffusion Model to Shrink Proteins While Maintaining Their Function
Nov 10, 2025Many proteins useful in modern medicine or bioengineering are challenging to make in the lab, fuse with other proteins in cells, or deliver to tissues in the body, because their sequences are too long. Shortening these sequences typically involves costly, time-consuming experimental campaigns. Ideally, we could instead use modern models of massive databases of sequences from nature to learn how to propose shrunken proteins that resemble sequences found in nature. Unfortunately, these models struggle to efficiently search the combinatorial space of all deletions, and are not trained with inductive biases to learn how to delete. To address this gap, we propose SCISOR, a novel discrete diffusion model that deletes letters from sequences to generate protein samples that resemble those found in nature. To do so, SCISOR trains a de-noiser to reverse a forward noising process that adds random insertions to natural sequences. As a generative model, SCISOR fits evolutionary sequence data competitively with previous large models. In evaluation, SCISOR achieves state-of-the-art predictions of the functional effects of deletions on ProteinGym. Finally, we use the SCISOR de-noiser to shrink long protein sequences, and show that its suggested deletions result in significantly more realistic proteins and more often preserve functional motifs than previous models of evolutionary sequences.
Protriever: End-to-End Differentiable Protein Homology Search for Fitness Prediction
Jun 10, 2025Retrieving homologous protein sequences is essential for a broad range of protein modeling tasks such as fitness prediction, protein design, structure modeling, and protein-protein interactions. Traditional workflows have relied on a two-step process: first retrieving homologs via Multiple Sequence Alignments (MSA), then training models on one or more of these alignments. However, MSA-based retrieval is computationally expensive, struggles with highly divergent sequences or complex insertions & deletions patterns, and operates independently of the downstream modeling objective. We introduce Protriever, an end-to-end differentiable framework that learns to retrieve relevant homologs while simultaneously training for the target task. When applied to protein fitness prediction, Protriever achieves state-of-the-art performance compared to sequence-based models that rely on MSA-based homolog retrieval, while being two orders of magnitude faster through efficient vector search. Protriever is both architecture- and task-agnostic, and can flexibly adapt to different retrieval strategies and protein databases at inference time -- offering a scalable alternative to alignment-centric approaches.
Multi-megabase scale genome interpretation with genetic language models
Jan 13, 2025Understanding how molecular changes caused by genetic variation drive disease risk is crucial for deciphering disease mechanisms. However, interpreting genome sequences is challenging because of the vast size of the human genome, and because its consequences manifest across a wide range of cells, tissues and scales -- spanning from molecular to whole organism level. Here, we present Phenformer, a multi-scale genetic language model that learns to generate mechanistic hypotheses as to how differences in genome sequence lead to disease-relevant changes in expression across cell types and tissues directly from DNA sequences of up to 88 million base pairs. Using whole genome sequencing data from more than 150 000 individuals, we show that Phenformer generates mechanistic hypotheses about disease-relevant cell and tissue types that match literature better than existing state-of-the-art methods, while using only sequence data. Furthermore, disease risk predictors enriched by Phenformer show improved prediction performance and generalisation to diverse populations. Accurate multi-megabase scale interpretation of whole genomes without additional experimental data enables both a deeper understanding of molecular mechanisms involved in disease and improved disease risk prediction at the level of individuals.
RITA: a Study on Scaling Up Generative Protein Sequence Models
May 11, 2022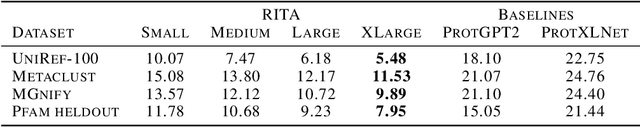
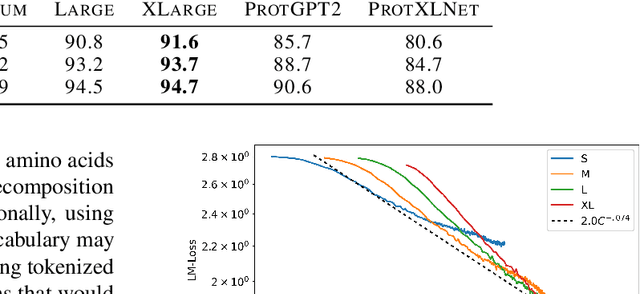
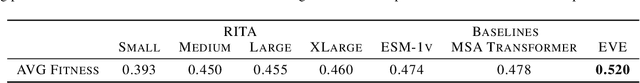
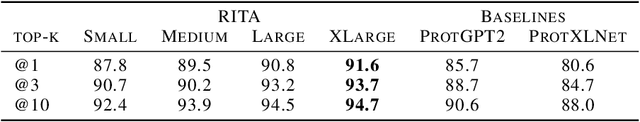
In this work we introduce RITA: a suite of autoregressive generative models for protein sequences, with up to 1.2 billion parameters, trained on over 280 million protein sequences belonging to the UniRef-100 database. Such generative models hold the promise of greatly accelerating protein design. We conduct the first systematic study of how capabilities evolve with model size for autoregressive transformers in the protein domain: we evaluate RITA models in next amino acid prediction, zero-shot fitness, and enzyme function prediction, showing benefits from increased scale. We release the RITA models openly, to the benefit of the research community.
Variational Inference for Sparse and Undirected Models
Jun 14, 2017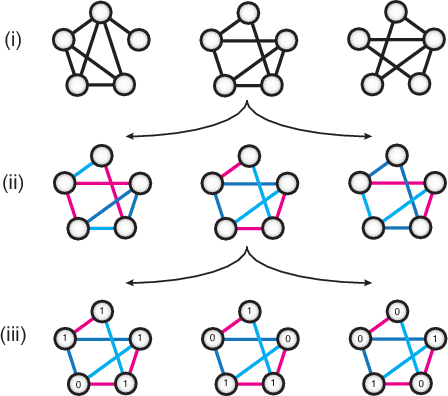

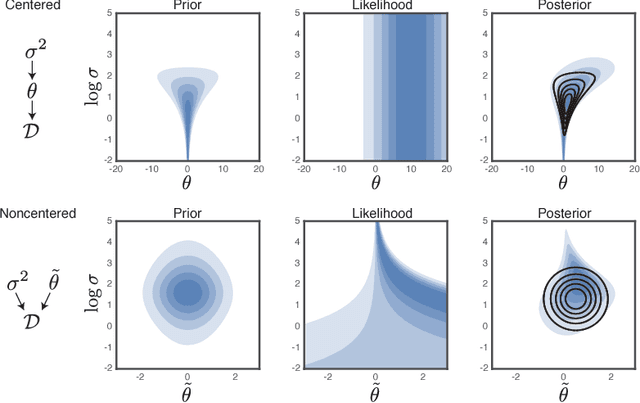

Undirected graphical models are applied in genomics, protein structure prediction, and neuroscience to identify sparse interactions that underlie discrete data. Although Bayesian methods for inference would be favorable in these contexts, they are rarely used because they require doubly intractable Monte Carlo sampling. Here, we develop a framework for scalable Bayesian inference of discrete undirected models based on two new methods. The first is Persistent VI, an algorithm for variational inference of discrete undirected models that avoids doubly intractable MCMC and approximations of the partition function. The second is Fadeout, a reparameterization approach for variational inference under sparsity-inducing priors that captures a posteriori correlations between parameters and hyperparameters with noncentered parameterizations. We find that, together, these methods for variational inference substantially improve learning of sparse undirected graphical models in simulated and real problems from physics and biology.