 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMemGraphRAG: Memory-based Multi-Agent System for Graph Retrieval-Augmented Generation
May 30, 2026Retrieval-Augmented Generation (RAG) has become an essential method for mitigating hallucinations in Large Language Models (LLMs) by leveraging external knowledge. Although effective for simple queries, traditional RAG struggles with large-scale, unstructured corpora where information is highly fragmented. Graph-based RAG (GraphRAG) incorporates knowledge graphs to capture structural relationships, enabling more comprehensive retrieval for complex reasoning. However, existing GraphRAG methods rely on isolated, fragment-level extraction for graph construction, lacking a global perspective on the whole corpus. As a result, these methods frequently lead to thematically inconsistent, logically conflicting, and structurally fragmented graphs that degrade retrieval performance. In this paper, we propose MemGraphRAG, a novel framework that introduces a memory-based multi-agent system to ensure high-quality graph construction. Specifically, MemGraphRAG employs a collaborative society of agents supported by shared memory, which provides a unified global context throughout the extraction process. This mechanism allows agents to dynamically resolve logical conflicts and maintain structural connectivity throughout the corpus. Furthermore, we propose a memory-aware hierarchical retrieval algorithm tailored for the constructed graph. Extensive experiments on multiple benchmarks demonstrate that MemGraphRAG outperforms the state-of-the-art baseline models with comparable efficiency. Our code is available at https://github.com/XMUDeepLIT/MemGraphRAG.
When to use Graphs in RAG: A Comprehensive Analysis for Graph Retrieval-Augmented Generation
Jun 06, 2025Graph retrieval-augmented generation (GraphRAG) has emerged as a powerful paradigm for enhancing large language models (LLMs) with external knowledge. It leverages graphs to model the hierarchical structure between specific concepts, enabling more coherent and effective knowledge retrieval for accurate reasoning.Despite its conceptual promise, recent studies report that GraphRAG frequently underperforms vanilla RAG on many real-world tasks. This raises a critical question: Is GraphRAG really effective, and in which scenarios do graph structures provide measurable benefits for RAG systems? To address this, we propose GraphRAG-Bench, a comprehensive benchmark designed to evaluate GraphRAG models onboth hierarchical knowledge retrieval and deep contextual reasoning. GraphRAG-Bench features a comprehensive dataset with tasks of increasing difficulty, coveringfact retrieval, complex reasoning, contextual summarization, and creative generation, and a systematic evaluation across the entire pipeline, from graph constructionand knowledge retrieval to final generation. Leveraging this novel benchmark, we systematically investigate the conditions when GraphRAG surpasses traditional RAG and the underlying reasons for its success, offering guidelines for its practical application. All related resources and analyses are collected for the community at https://github.com/GraphRAG-Bench/GraphRAG-Benchmark.
Schrödinger-ANI: An Eight-Element Neural Network Interaction Potential with Greatly Expanded Coverage of Druglike Chemical Space
Nov 22, 2019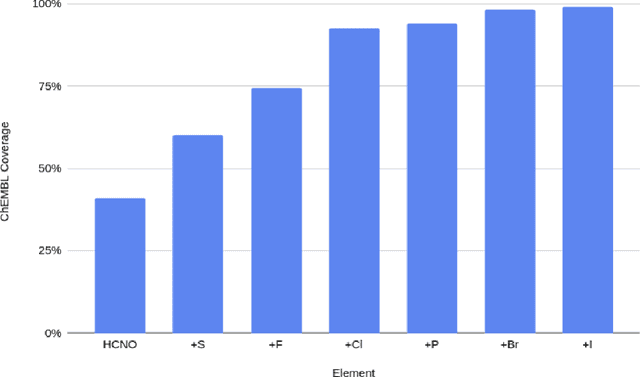
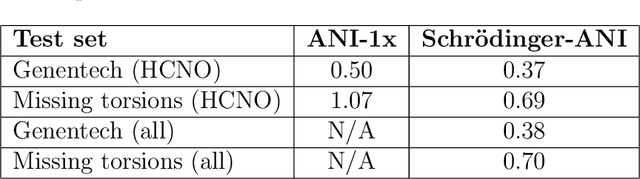
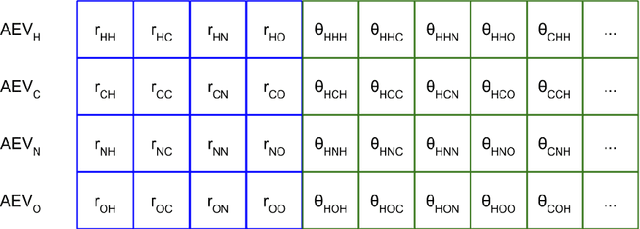
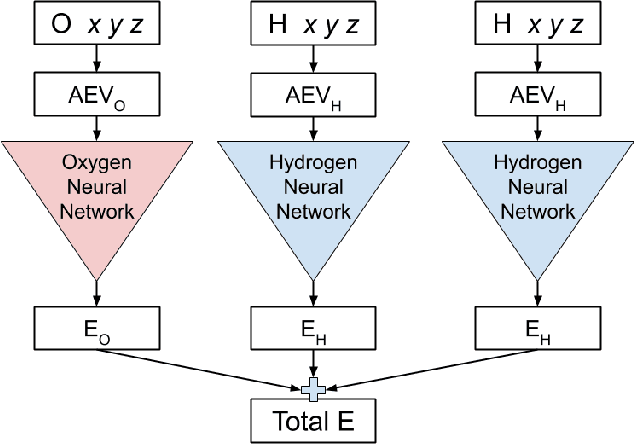
We have developed a neural network potential energy function for use in drug discovery, with chemical element support extended from 41% to 94% of druglike molecules based on ChEMBL. We expand on the work of Smith et al., with their highly accurate network for the elements H, C, N, O, creating a network for H, C, N, O, S, F, Cl, P. We focus particularly on the calculation of relative conformer energies, for which we show that our new potential energy function has an RMSE of 0.70 kcal/mol for prospective druglike molecule conformers, substantially better than the previous state of the art. The speed and accuracy of this model could greatly accelerate the parameterization of protein-ligand binding free energy calculations for novel druglike molecules.