 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSTRAND: Sequence-Conditioned Transport for Single-Cell Perturbations
Feb 10, 2026Predicting how genetic perturbations change cellular state is a core problem for building controllable models of gene regulation. Perturbations targeting the same gene can produce different transcriptional responses depending on their genomic locus, including different transcription start sites and regulatory elements. Gene-level perturbation models collapse these distinct interventions into the same representation. We introduce STRAND, a generative model that predicts single-cell transcriptional responses by conditioning on regulatory DNA sequence. STRAND represents a perturbation by encoding the sequence at its genomic locus and uses this representation to parameterize a conditional transport process from control to perturbed cell states. Representing perturbations by sequence, rather than by a fixed set of gene identifiers, supports zero-shot inference at loci not seen during training and expands inference-time genomic coverage from ~1.5% for gene-level single-cell foundation models to ~95% of the genome. We evaluate STRAND on CRISPR perturbation datasets in K562, Jurkat, and RPE1 cells. STRAND improves discrimination scores by up to 33% in low-sample regimes, achieves the best average rank on unseen gene perturbation benchmarks, and improves transfer to novel cell lines by up to 0.14 in Pearson correlation. Ablations isolate the gains to sequence conditioning and transport, and case studies show that STRAND resolves functionally alternative transcription start sites missed by gene-level models.
Marginal Contribution Feature Importance -- an Axiomatic Approach for The Natural Case
Oct 15, 2020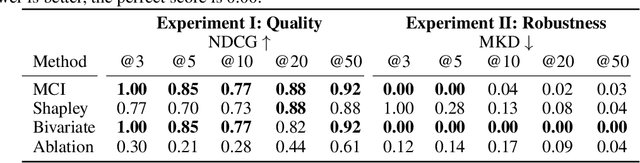
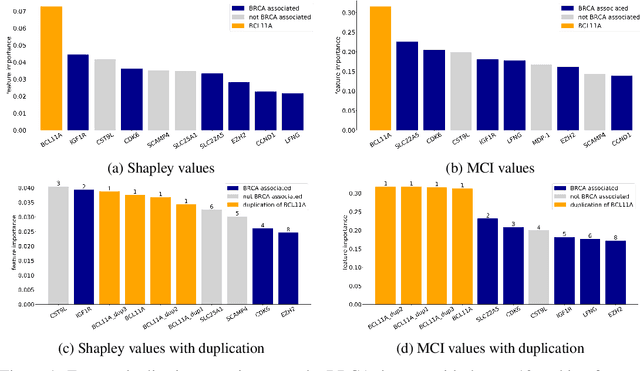
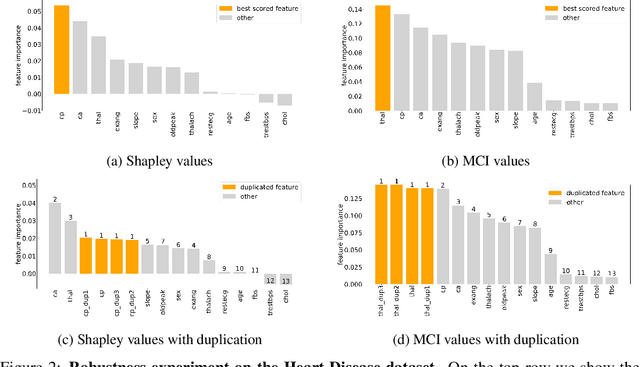
When training a predictive model over medical data, the goal is sometimes to gain insights about a certain disease. In such cases, it is common to use feature importance as a tool to highlight significant factors contributing to that disease. As there are many existing methods for computing feature importance scores, understanding their relative merits is not trivial. Further, the diversity of scenarios in which they are used lead to different expectations from the feature importance scores. While it is common to make the distinction between local scores that focus on individual predictions and global scores that look at the contribution of a feature to the model, another important division distinguishes model scenarios, in which the goal is to understand predictions of a given model from natural scenarios, in which the goal is to understand a phenomenon such as a disease. We develop a set of axioms that represent the properties expected from a feature importance function in the natural scenario and prove that there exists only one function that satisfies all of them, the Marginal Contribution Feature Importance (MCI). We analyze this function for its theoretical and empirical properties and compare it to other feature importance scores. While our focus is the natural scenario, we suggest that our axiomatic approach could be carried out in other scenarios too.